

Publications
85 Quantum nuclear dynamics on a distributed set of ion-trap quantum computing systems
Anurag Dwivedi, A. J. Rasmusson, Philip Richerme, Srinivasan S. Iyengar, "Quantum nuclear dynamics on a distributed set of ion-trap quantum computing systems", J. Am. Chem. Soc. ASAP (2024).
84 Resource optimization for quantum dynamics with tensor networks: Quantum and Classical Algorithms
A. Dwivedi, M. A. Lopez-Ruiz, and S. S. Iyengar, “Resource optimization for quantum dynamics with tensor networks: Quantum and Classical Algorithms” J. Phys. Chem. A, 128 , 6774 (2024).
83 Rare Events Sampling methods for Quantum and Classical ab initio Molecular Dynamics
Srinivasan S. Iyengar, H. Bernhard Schlegel, Isaiah Sumner, Junjie Li, “Rare Events Sampling methods for Quantum and Classical ab initio Molecular Dynamics” J. Phys. Chem. A , 128, 5386 (2024).
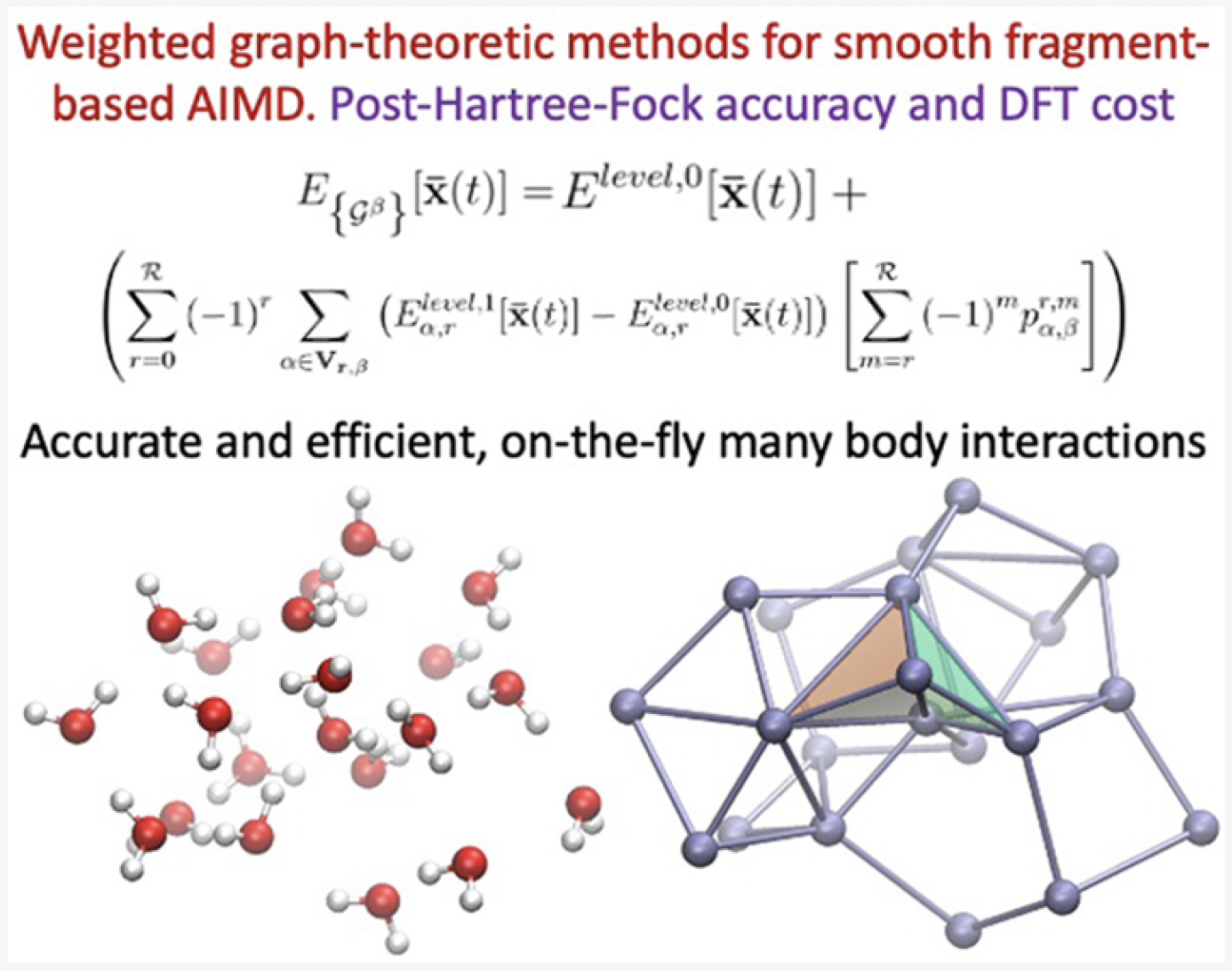
82 A reformulation of all ONIOM-type molecular fragmentation approaches using graph theory-based projection operators: Applications to dynamics, molecular potential surfaces, and machine learning and quantum computing
S. S. Iyengar, Timothy C. Ricard, Xiao Zhu, “A reformulation of all ONIOM-type molecular fragmentation approaches using graph theory-based projection operators: Applications to dynamics, molecular potential surfaces, and machine learning and quantum computing” J. Phys. Chem. A, ASAP (2023). J. Phys. Chem. A 128, 466 (2024).
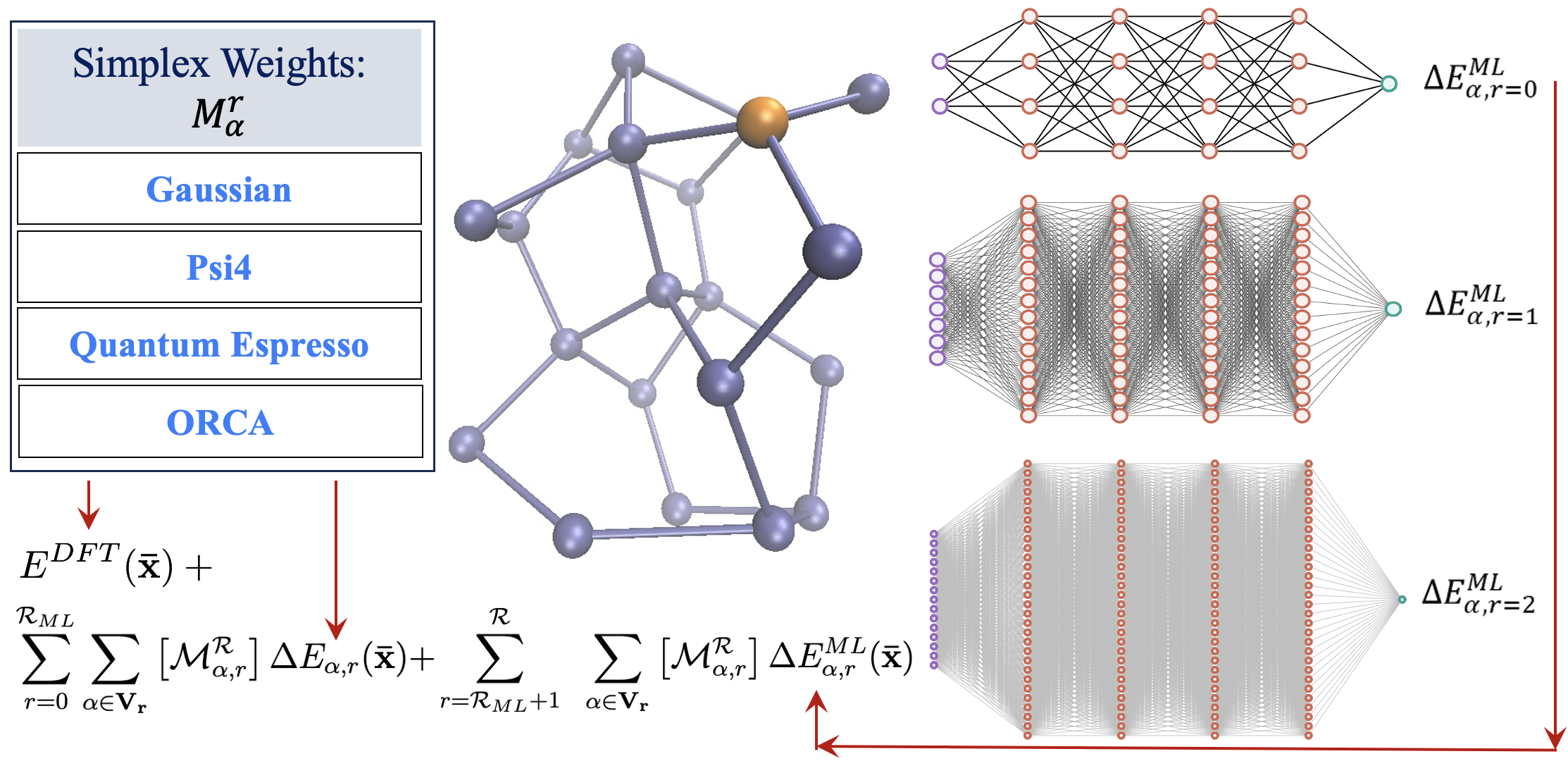 Summary: We present a graph-theory-based reformulation of all ONIOM-based molecular fragmentation methods. We discuss applications to (a) accurate post-Hartree–Fock AIMD that can be conducted at DFT cost for medium-sized systems, (b) hybrid DFT condensed-phase studies at the cost of pure density functionals, (c) reduced cost on-the-fly large basis gas-phase AIMD and condensed-phase studies, (d) post-Hartree–Fock-level potential surfaces at DFT cost to obtain quantum nuclear effects, and (e) novel transfer machine learning protocols derived from these measures. Additionally, in previous work, the unifying strategy discussed here has been used to construct new quantum computing algorithms. Thus, we conclude that this reformulation is robust and accurate.
Summary: We present a graph-theory-based reformulation of all ONIOM-based molecular fragmentation methods. We discuss applications to (a) accurate post-Hartree–Fock AIMD that can be conducted at DFT cost for medium-sized systems, (b) hybrid DFT condensed-phase studies at the cost of pure density functionals, (c) reduced cost on-the-fly large basis gas-phase AIMD and condensed-phase studies, (d) post-Hartree–Fock-level potential surfaces at DFT cost to obtain quantum nuclear effects, and (e) novel transfer machine learning protocols derived from these measures. Additionally, in previous work, the unifying strategy discussed here has been used to construct new quantum computing algorithms. Thus, we conclude that this reformulation is robust and accurate.
81 Capturing weak interactions in surface adsorbate systems at coupled cluster accuracy: a graph-theoretic molecular fragmentation approach improved through machine learning
Timothy C. Ricard, Xiao Zhu, and S. S. Iyengar, “Capturing weak interactions in surface adsorbate systems at coupled cluster accuracy: a graph-theoretic molecular fragmentation approach improved through machine learning”. J. Chem. Theory and Comput. 19, 8541 (2023).
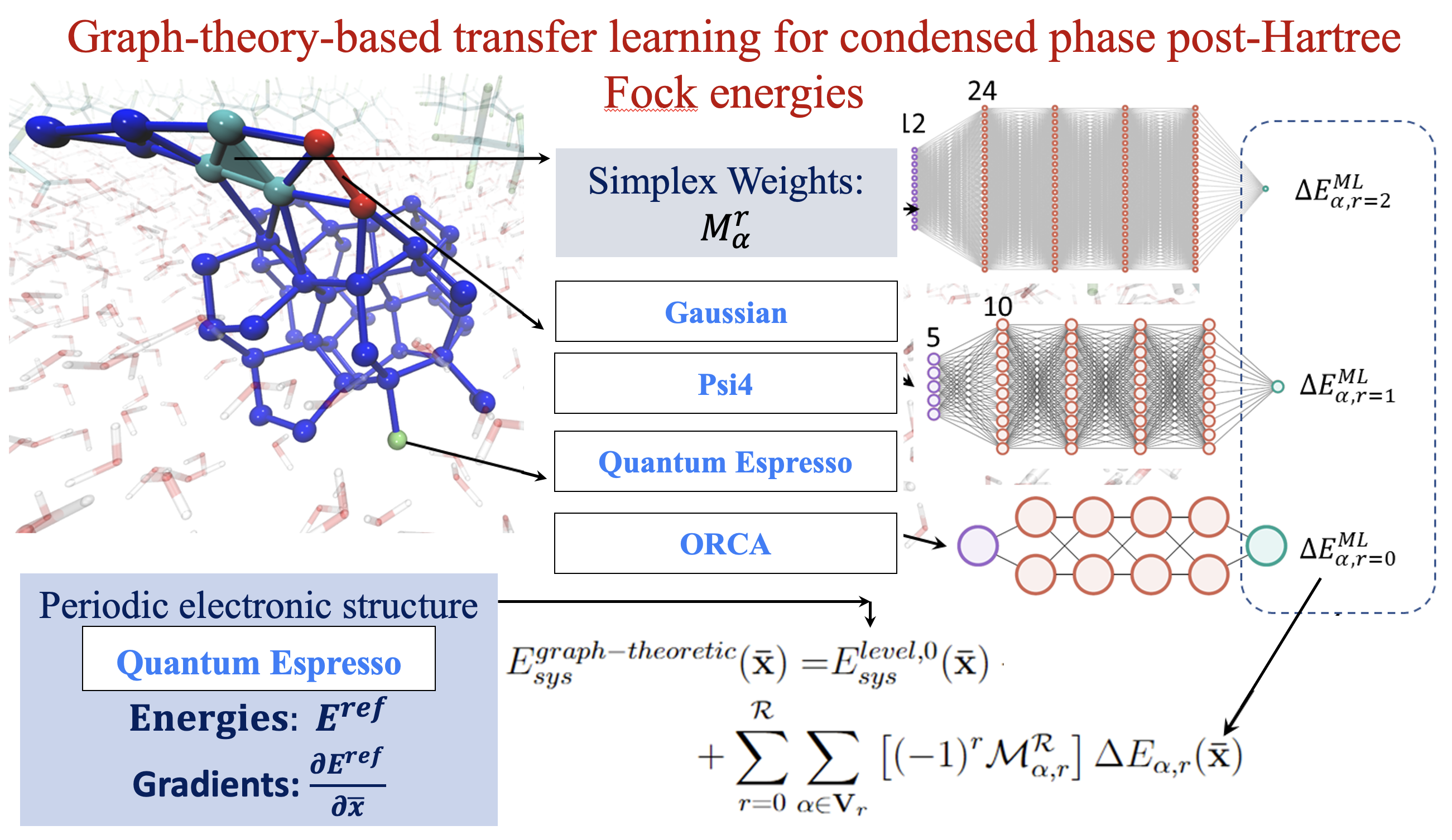 Summary: The accurate and efficient study of the interactions of organic matter with the surface of water is critical to a wide range of applications. For example, environmental studies have found that acidic polyfluorinated alkyl substances, especially perfluorooctanoic acid (PFOA), have spread throughout the environment and bioaccumulate into human populations residing near contaminated watersheds, leading to many systemic maladies. Thus, the study of the interactions of PFOA with water surfaces became important for the mitigation of their activity as pollutants and threats to public health. However, theoretical study of the interactions of such organic adsorbates on the surface of water, and their bulk concerted properties, often necessitates the use of ab initio methods to properly incorporate the long-range electronic properties that govern these extended systems. Notable theoretical treatments of “on-water” reactions thus far have employed hybrid DFT and semilocal DFT, but the interactions involved are weak interactions that may be best described using post-Hartree–Fock theory. Here, we aim to demonstrate the utility of a graph-theoretic approach to molecular fragmentation that accurately captures the critical “weak” interactions while maintaining an efficient ab initio treatment of the long-range periodic interactions that underpin the physics of extended systems. We apply this graph-theoretical treatment to study PFOA on the surface of water as a model system for the study of weak interactions seen in the wide range of surface interactions and reactions. The approach divides a system into a set of vertices, that are then connected through edges, faces, and higher order graph theoretic objects known as simplexes, to represent a collection of locally interacting subsystems. These subsystems are then used to construct ab initio molecular dynamics simulations and for computing multidimensional potential energy surfaces. To further improve the computational efficiency of our graph theoretic fragmentation method, we use a recently developed transfer learning protocol to construct the full system potential energy from a family of neural networks each designed to accurately model the behavior of individual simplexes. We use a unique multidimensional clustering algorithm, based on the k-means clustering methodology, to define our training space for each separate simplex. These models are used to extrapolate the energies for molecular dynamics trajectories at PFOA water interfaces, at less than one-tenth the cost as compared to a regular molecular fragmentation-based dynamics calculation with excellent agreement with couple cluster level of full system potential energies.
Summary: The accurate and efficient study of the interactions of organic matter with the surface of water is critical to a wide range of applications. For example, environmental studies have found that acidic polyfluorinated alkyl substances, especially perfluorooctanoic acid (PFOA), have spread throughout the environment and bioaccumulate into human populations residing near contaminated watersheds, leading to many systemic maladies. Thus, the study of the interactions of PFOA with water surfaces became important for the mitigation of their activity as pollutants and threats to public health. However, theoretical study of the interactions of such organic adsorbates on the surface of water, and their bulk concerted properties, often necessitates the use of ab initio methods to properly incorporate the long-range electronic properties that govern these extended systems. Notable theoretical treatments of “on-water” reactions thus far have employed hybrid DFT and semilocal DFT, but the interactions involved are weak interactions that may be best described using post-Hartree–Fock theory. Here, we aim to demonstrate the utility of a graph-theoretic approach to molecular fragmentation that accurately captures the critical “weak” interactions while maintaining an efficient ab initio treatment of the long-range periodic interactions that underpin the physics of extended systems. We apply this graph-theoretical treatment to study PFOA on the surface of water as a model system for the study of weak interactions seen in the wide range of surface interactions and reactions. The approach divides a system into a set of vertices, that are then connected through edges, faces, and higher order graph theoretic objects known as simplexes, to represent a collection of locally interacting subsystems. These subsystems are then used to construct ab initio molecular dynamics simulations and for computing multidimensional potential energy surfaces. To further improve the computational efficiency of our graph theoretic fragmentation method, we use a recently developed transfer learning protocol to construct the full system potential energy from a family of neural networks each designed to accurately model the behavior of individual simplexes. We use a unique multidimensional clustering algorithm, based on the k-means clustering methodology, to define our training space for each separate simplex. These models are used to extrapolate the energies for molecular dynamics trajectories at PFOA water interfaces, at less than one-tenth the cost as compared to a regular molecular fragmentation-based dynamics calculation with excellent agreement with couple cluster level of full system potential energies.
80 Graph-|Q⟩⟨C|: A Quantum algorithm with Reduced quantum circuit depth for electronic structure
S. S. Iyengar, Juncheng (Harry) Zhang, Timothy C. Ricard and Debadrita Saha, “Graph-|Q⟩⟨C|: A Quantum algorithm with Reduced quantum circuit depth for electronic structure”, J. Phys. Chem. A, 127, 9334 (2023).
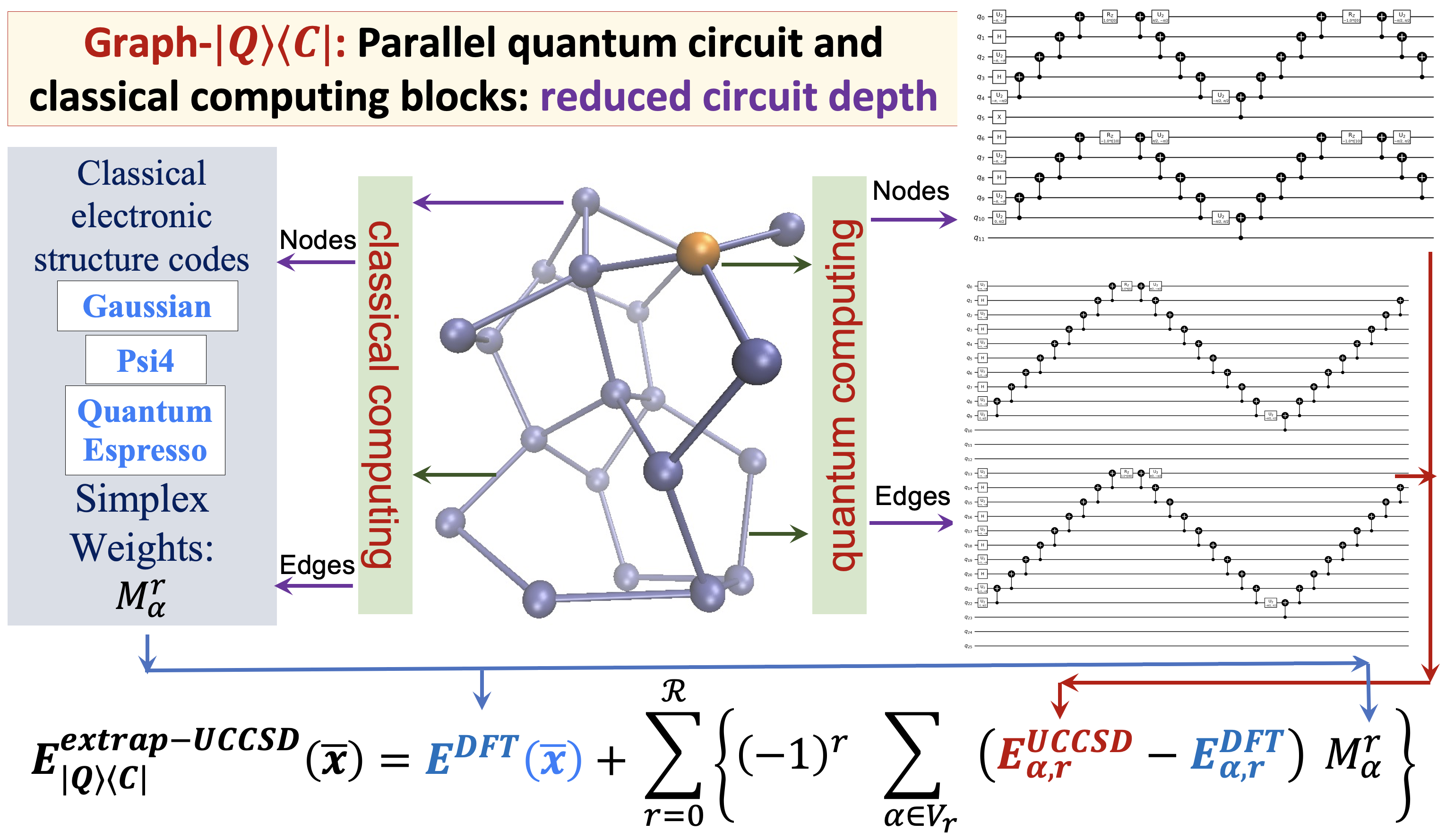 Summary: The accurate determination of chemical properties is known to have a critical impact on multiple fundamental chemical problems but is deeply hindered by the steep algebraic scaling of electron correlation calculations and the exponential scaling of quantum nuclear dynamics. With the advent of new quantum computing hardware and associated developments in creating new paradigms for quantum software, this avenue has been recognized as perhaps one way to address exponentially complex challenges in quantum chemistry and molecular dynamics. In this paper, we discuss a new approach to drastically reduce the quantum circuit depth (by several orders of magnitude) and help improve the accuracy in the quantum computation of electron correlation energies for large molecular systems. The method is derived from a graph-theoretic approach to molecular fragmentation and enables us to create a family of projection operators that decompose quantum circuits into separate unitary processes. Some of these processes can be treated on quantum hardware and others on classical hardware in a completely asynchronous and parallel fashion. Numerical benchmarks are provided through the computation of unitary coupled-cluster singles and doubles (UCCSD) energies for medium-sized protonated and neutral water clusters using the new quantum algorithms presented here.
Summary: The accurate determination of chemical properties is known to have a critical impact on multiple fundamental chemical problems but is deeply hindered by the steep algebraic scaling of electron correlation calculations and the exponential scaling of quantum nuclear dynamics. With the advent of new quantum computing hardware and associated developments in creating new paradigms for quantum software, this avenue has been recognized as perhaps one way to address exponentially complex challenges in quantum chemistry and molecular dynamics. In this paper, we discuss a new approach to drastically reduce the quantum circuit depth (by several orders of magnitude) and help improve the accuracy in the quantum computation of electron correlation energies for large molecular systems. The method is derived from a graph-theoretic approach to molecular fragmentation and enables us to create a family of projection operators that decompose quantum circuits into separate unitary processes. Some of these processes can be treated on quantum hardware and others on classical hardware in a completely asynchronous and parallel fashion. Numerical benchmarks are provided through the computation of unitary coupled-cluster singles and doubles (UCCSD) energies for medium-sized protonated and neutral water clusters using the new quantum algorithms presented here.
79 A Synthesis of Hidden Subgroup Quantum Algorithms and Quantum Chemical Dynamics
S. S. Iyengar, A. Kumar, Debadrita Saha, and A. Sabry, “A Synthesis of Hidden Subgroup Quantum Algorithms and Quantum Chemical Dynamics”. J. Chem. Theory and Comput. 19, 6082 (2023).
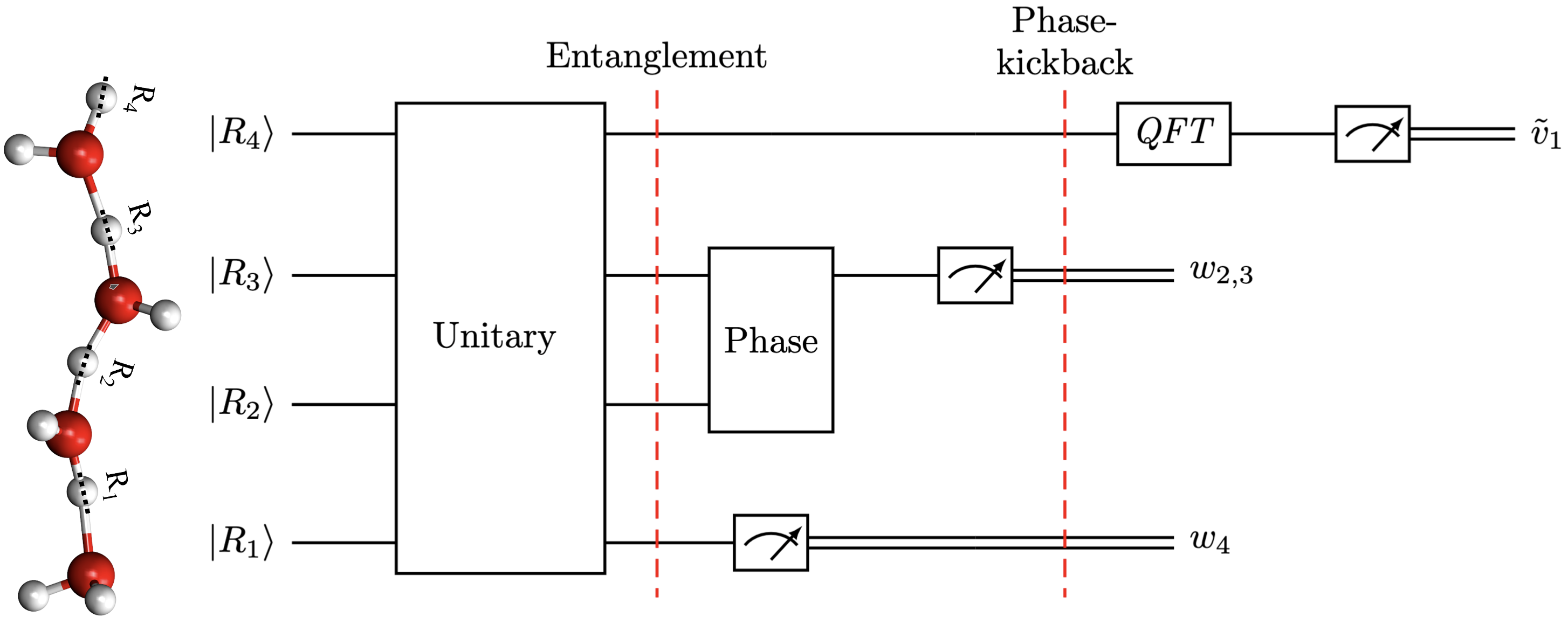 Summary: We describe a general formalism for quantum dynamics and show how this formalism subsumes several quantum algorithms including the Deutsch, Deutsch-Jozsa, Bernstein-Vazirani, Simon, and Shor algorithms as well as the conventional approach to quantum dynamics based on tensor networks. The common framework exposes similarities among quantum algorithms and natural quantum phenomena: we illustrate this connection by showing how the correlated behavior of protons in water wire systems that are common in many biological and materials systems parallels the structure of Shor's algorithm.
Summary: We describe a general formalism for quantum dynamics and show how this formalism subsumes several quantum algorithms including the Deutsch, Deutsch-Jozsa, Bernstein-Vazirani, Simon, and Shor algorithms as well as the conventional approach to quantum dynamics based on tensor networks. The common framework exposes similarities among quantum algorithms and natural quantum phenomena: we illustrate this connection by showing how the correlated behavior of protons in water wire systems that are common in many biological and materials systems parallels the structure of Shor's algorithm.
78 Quantum Computing with Dartboards
Ishaan Ganti , and S. S. Iyengar, “Quantum Computing with Dartboards” J. Phys. Chem. A, 127 , 7853 (2023).
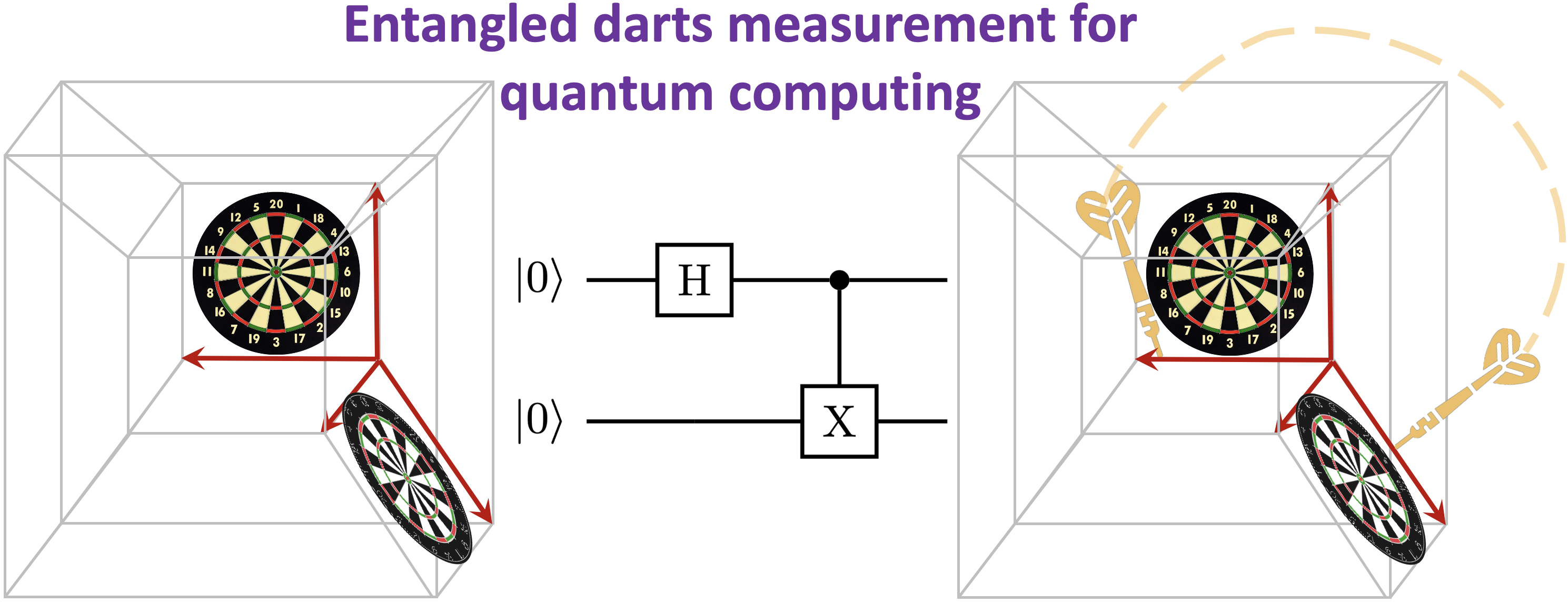 Summary: We present a physically appealing and elegant picture for quantum computing using rules constructed for a game of darts. A dartboard is used to represent the state space in quantum mechanics and the act of throwing the dart is shown to have close similarities to the concept of measurement, or collapse of the wavefunction in quantum mechanics. The analogy is constructed in arbitrary dimensional spaces, that is using arbitrary dimensional dartboards, and for for such arbitrary spaces this also provides us a ``visual'' description of uncertainty. Finally, connections of qubits and quantum computing algorithms is also made opening the possibility to construct analogies between quantum algorithms and coupled dart-throw competitions.
Summary: We present a physically appealing and elegant picture for quantum computing using rules constructed for a game of darts. A dartboard is used to represent the state space in quantum mechanics and the act of throwing the dart is shown to have close similarities to the concept of measurement, or collapse of the wavefunction in quantum mechanics. The analogy is constructed in arbitrary dimensional spaces, that is using arbitrary dimensional dartboards, and for for such arbitrary spaces this also provides us a ``visual'' description of uncertainty. Finally, connections of qubits and quantum computing algorithms is also made opening the possibility to construct analogies between quantum algorithms and coupled dart-throw competitions.
77 Quantum Computation of Hydrogen Bond Dynamics and Vibrational Spectra
P. Richerme, Melissa C. Revelle, Debadrita Saha, Miguel Angel Lopez-Ruiz, Anurag Dwivedi, Sam A. Norrell, Christopher G. Yale, Daniel Lobser, Ashlyn D. Burch, Susan M. Clark, J. M. Smith, A. Sabry, and S. S. Iyengar “Quantum Computation of Hydrogen Bond Dynamics and Vibrational Spectra”. J. Phys. Chem. Lett. 14, 7256 (2023).
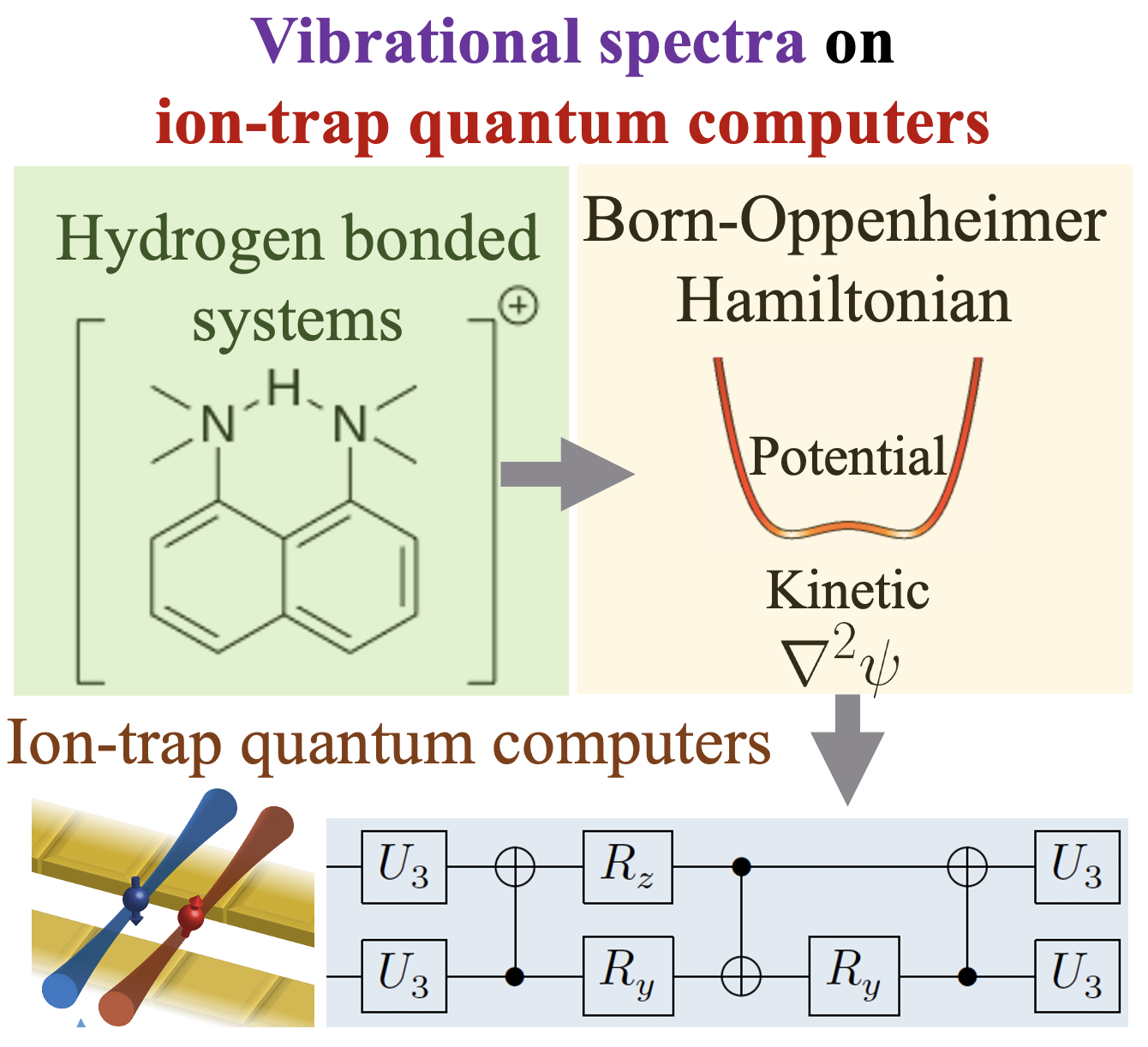 Summary: Calculating observable properties of chemical systems is often classically intractable and widely viewed as a promising application of quantum information processing. Here, we introduce a new framework for solving generic quantum chemical dynamics problems using quantum logic. We experimentally demonstrate a proof-of-principle instance of our method using the QSCOUT ion-trap quantum computer, where we experimentally drive the ion-trap system to emulate the quantum wavepacket dynamics corresponding to the shared-proton within an anharmonic hydrogen bonded system. Following the experimental creation and propagation of the shared-proton wavepacket on the ion-trap, we extract measurement observables such as its time-dependent spatial projection and its characteristic vibrational frequencies to spectroscopicaccuracy (3.3 cm−1 wavenumbers, corresponding to >99.9% fidelity). Our approach introduces a new paradigm for studying the chemical dynamics and vibrational spectra of molecules and opens the possibility to describe the behavior of complex molecular processes with unprecedented accuracy.
Summary: Calculating observable properties of chemical systems is often classically intractable and widely viewed as a promising application of quantum information processing. Here, we introduce a new framework for solving generic quantum chemical dynamics problems using quantum logic. We experimentally demonstrate a proof-of-principle instance of our method using the QSCOUT ion-trap quantum computer, where we experimentally drive the ion-trap system to emulate the quantum wavepacket dynamics corresponding to the shared-proton within an anharmonic hydrogen bonded system. Following the experimental creation and propagation of the shared-proton wavepacket on the ion-trap, we extract measurement observables such as its time-dependent spatial projection and its characteristic vibrational frequencies to spectroscopicaccuracy (3.3 cm−1 wavenumbers, corresponding to >99.9% fidelity). Our approach introduces a new paradigm for studying the chemical dynamics and vibrational spectra of molecules and opens the possibility to describe the behavior of complex molecular processes with unprecedented accuracy.
76 Quantum algorithms for the study of electronic structure and molecular dynamics: Novel computational protocols
S. S. Iyengar, Debadrita Saha, Anurag Dwivedi, Miguel Angel Lopez-Ruiz, Anup Kumar, Juncheng (Harry) Zhang, Timothy C. Ricard, Philip Richerme Amr Sabry “Quantum algorithms for the study of electronic structure and molecular dynamics: Novel computational protocols” In Comprehensive Computational Chemistry. In Press. (2023)
Summary: The accurate computational determination of chemical, materials, biological, and atmospheric properties has critical impact on a wide range of health and environmental problems, but is deeply limited by the computational scaling of quantum-mechanical methods. The complexity of quantum-chemical studies arises from the steep algebraic scaling of electron correlation methods, and the exponential scaling in studying nuclear dynamics and molecular flexibility. In this article we provide an overview of the challenges involved in performing accurate post-Hartree-Fock electronic structure and quantum nuclear dynamics calculations on quantum hardware. For electronic structure, we present a procedure to drastically reduce the depth of quantum circuits and improve the accuracy of results in computing post-Hartree-Fock electronic structure energies for large molecular systems. The method is based on molecular fragmentation where a molecular system is divided into overlapping fragments through a graph theoretic procedure. This allows us to create a set of projection operators that decompose the unitary evolution of the full system into separate sets of unitary processes, some of which can be treated on quantum hardware and others on classical hardware. Thus, we develop a procedure for electronic structure that can be asynchronously spawned onto a potentially large ensemble of classical and quantum hardware systems. We also discuss a framework which allows for the solution of quantum chemical nuclear dynamics by mapping these to quantum spin-lattice simulators. This mapping procedure allows us to determine the local fields and spin-spin couplings needed to identically match the molecular and spin-lattice Hamiltonians and hence the resultant dynamics.
75 Exploiting analogies between Boltzmann machines and Feynman path integrals
S. S. Iyengar, and S. Kais “Exploiting analogies between Boltzmann machines and Feynman path integrals”. J. Chem. Theory and Comput. 19, 2446 (2023).
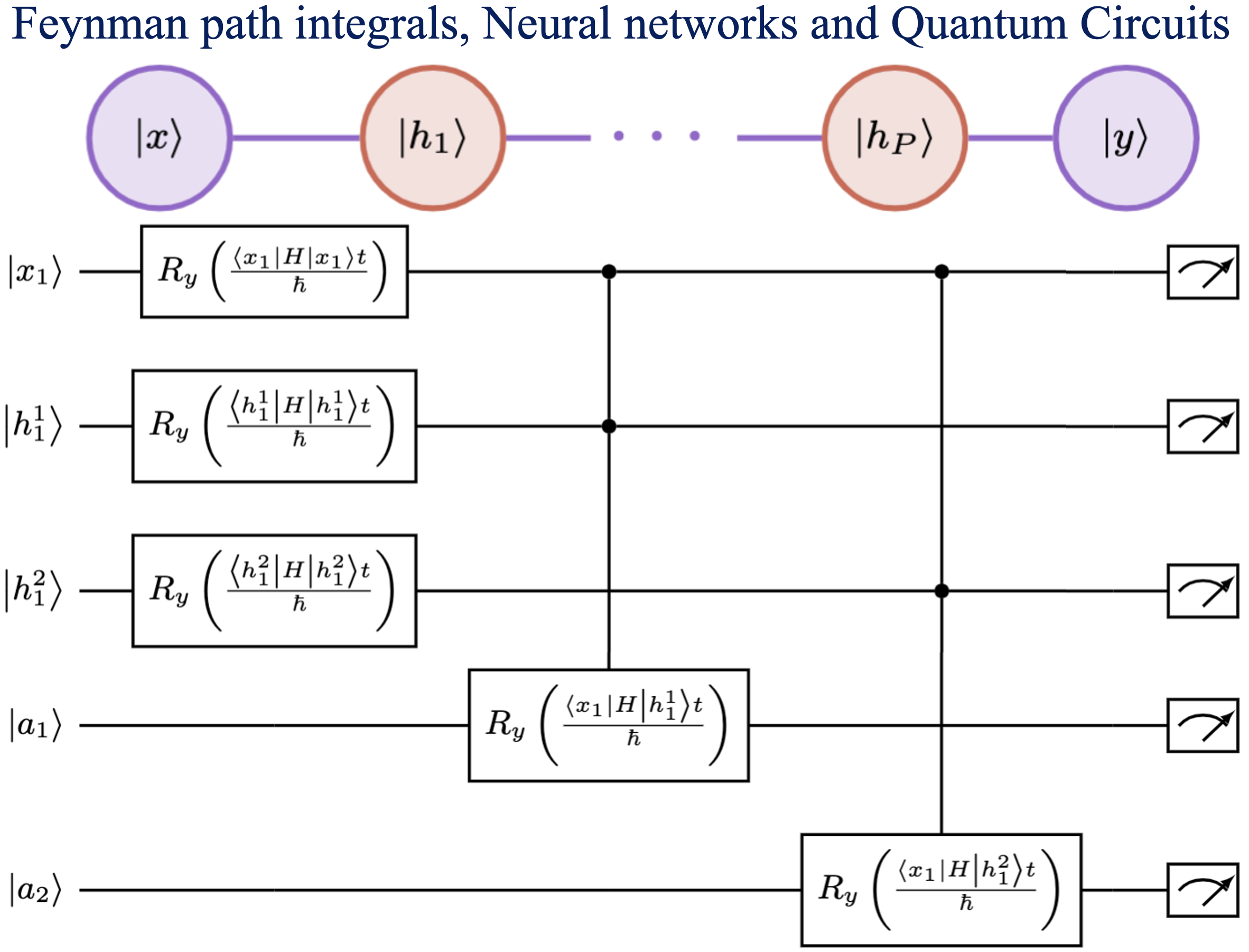 Summary: Machine learning has had a significant impact on multiple areas of science, technology, health, and computer and information sciences. Through the advent of quantum computing, quantum machine learning has developed as a new and important avenue for the study of complex learning problems. Yet there is substantial debate and uncertainty in regard to the foundations of machine learning. Here, we provide a detailed exposition of the mathematical connections between a general machine learning approach called Boltzmann machines and Feynman’s description of quantum and statistical mechanics. In Feynman’s description, quantum phenomena arise from an elegant, weighted sum over (or superposition of) paths. Our analysis shows that Boltzmann machines and neural networks have a similar mathematical structure. This allows the interpretation that the hidden layers in Boltzmann machines and neural networks are discrete versions of path elements and allows a path integral interpretation of machine learning similar to that in quantum and statistical mechanics. Since Feynman paths are a natural and elegant depiction of interference phenomena and the superposition principle germane to quantum mechanics, this analysis allows us to interpret the goal in machine learning as finding an appropriate combination of paths, and accumulated path-weights, through a network, that cumulatively captures the correct properties of an x-to-y map for a given mathematical problem. We are forced to conclude that neural networks are naturally related to Feynman path-integrals and hence may present one avenue to be considered as quantum problems. Consequently, we provide general quantum circuit models applicable to both Boltzmann machines and Feynman path integrals.
Summary: Machine learning has had a significant impact on multiple areas of science, technology, health, and computer and information sciences. Through the advent of quantum computing, quantum machine learning has developed as a new and important avenue for the study of complex learning problems. Yet there is substantial debate and uncertainty in regard to the foundations of machine learning. Here, we provide a detailed exposition of the mathematical connections between a general machine learning approach called Boltzmann machines and Feynman’s description of quantum and statistical mechanics. In Feynman’s description, quantum phenomena arise from an elegant, weighted sum over (or superposition of) paths. Our analysis shows that Boltzmann machines and neural networks have a similar mathematical structure. This allows the interpretation that the hidden layers in Boltzmann machines and neural networks are discrete versions of path elements and allows a path integral interpretation of machine learning similar to that in quantum and statistical mechanics. Since Feynman paths are a natural and elegant depiction of interference phenomena and the superposition principle germane to quantum mechanics, this analysis allows us to interpret the goal in machine learning as finding an appropriate combination of paths, and accumulated path-weights, through a network, that cumulatively captures the correct properties of an x-to-y map for a given mathematical problem. We are forced to conclude that neural networks are naturally related to Feynman path-integrals and hence may present one avenue to be considered as quantum problems. Consequently, we provide general quantum circuit models applicable to both Boltzmann machines and Feynman path integrals.
74 Graph-theoretic Molecular fragmentation for potential surfaces leads naturally to a tensor network form and allows accurate and efficient quantum nuclear dynamics
A. Kumar, N. DeGregorio T. C. Ricard, and S. S. Iyengar, “Graph-theoretic Molecular fragmentation for potential surfaces leads naturally to a tensor network form and allows accurate and efficient quantum nuclear dynamics”. J. Chem. Theory and Comput. 18, 7243 (2022).
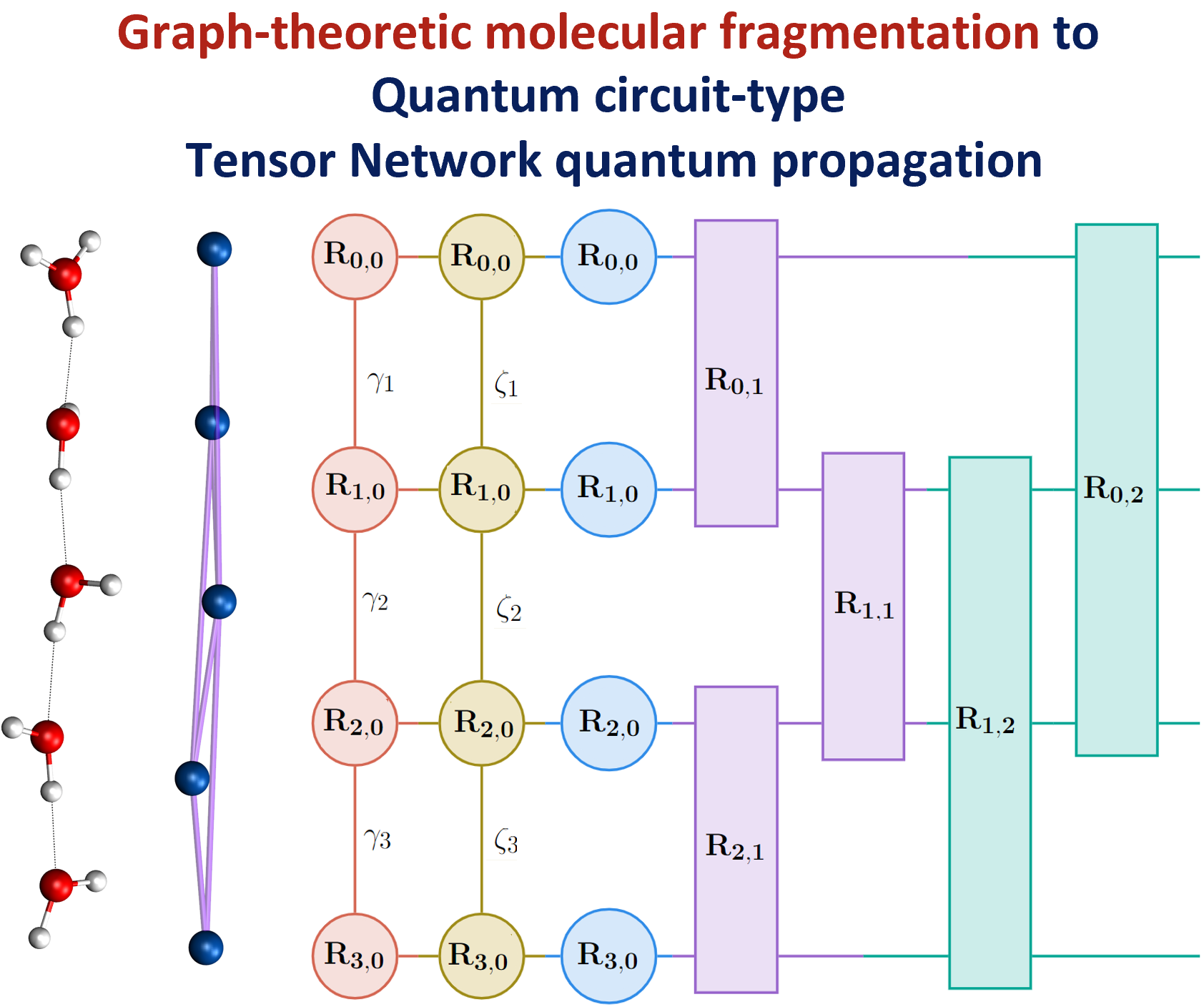 Summary: Molecular fragmentation methods have revolutionized quantum chemistry. Here, we use a graph-theoretically generated molecular fragmentation method, to obtain accurate and efficient representations for multidimensional potential energy surfaces and the quantum time-evolution operator, which plays a critical role in quantum chemical dynamics. In doing so, we find that the graph-theoretic fragmentation approach naturally reduces the potential portion of the time-evolution operator into a tensor network that contains a stream of coupled lower-dimensional propagation steps to potentially achieve quantum dynamics with reduced complexity. Furthermore, the fragmentation approach used here has previously been shown to allow accurate and efficient computation of post- Hartree−Fock electronic potential energy surfaces, which in many cases has been shown to be at density functional theory cost. Thus, by combining the advantages of molecular fragmentation with the tensor network formalism, the approach yields an on-the-fly quantum dynamics scheme where both the electronic potential calculation and nuclear propagation portion are enormously simplified through a single stroke. The method is demonstrated by computing approximations to the propagator and to potential surfaces for a set of coupled nuclear dimensions within a protonated water wire problem exhibiting the Grotthuss mechanism of proton transport. In all cases, our approach has been shown to reduce the complexity of representing the quantum propagator, and by extension action of the propagator on an initial wavepacket, by several orders, with minimal loss in accuracy.
Summary: Molecular fragmentation methods have revolutionized quantum chemistry. Here, we use a graph-theoretically generated molecular fragmentation method, to obtain accurate and efficient representations for multidimensional potential energy surfaces and the quantum time-evolution operator, which plays a critical role in quantum chemical dynamics. In doing so, we find that the graph-theoretic fragmentation approach naturally reduces the potential portion of the time-evolution operator into a tensor network that contains a stream of coupled lower-dimensional propagation steps to potentially achieve quantum dynamics with reduced complexity. Furthermore, the fragmentation approach used here has previously been shown to allow accurate and efficient computation of post- Hartree−Fock electronic potential energy surfaces, which in many cases has been shown to be at density functional theory cost. Thus, by combining the advantages of molecular fragmentation with the tensor network formalism, the approach yields an on-the-fly quantum dynamics scheme where both the electronic potential calculation and nuclear propagation portion are enormously simplified through a single stroke. The method is demonstrated by computing approximations to the propagator and to potential surfaces for a set of coupled nuclear dimensions within a protonated water wire problem exhibiting the Grotthuss mechanism of proton transport. In all cases, our approach has been shown to reduce the complexity of representing the quantum propagator, and by extension action of the propagator on an initial wavepacket, by several orders, with minimal loss in accuracy.
73 Graph theoretic molecular fragmentation methods for electronic potential energy surfaces augmented by machine learning
Xiao Zhu and S. S. Iyengar, “Graph Theoretic Molecular Fragmentation for Multidimensional Potential Energy Surfaces Yield an Adaptive and General Transfer Machine Learning Protocol”, Chem. Theory and Comput. 18, 5125 (2022).
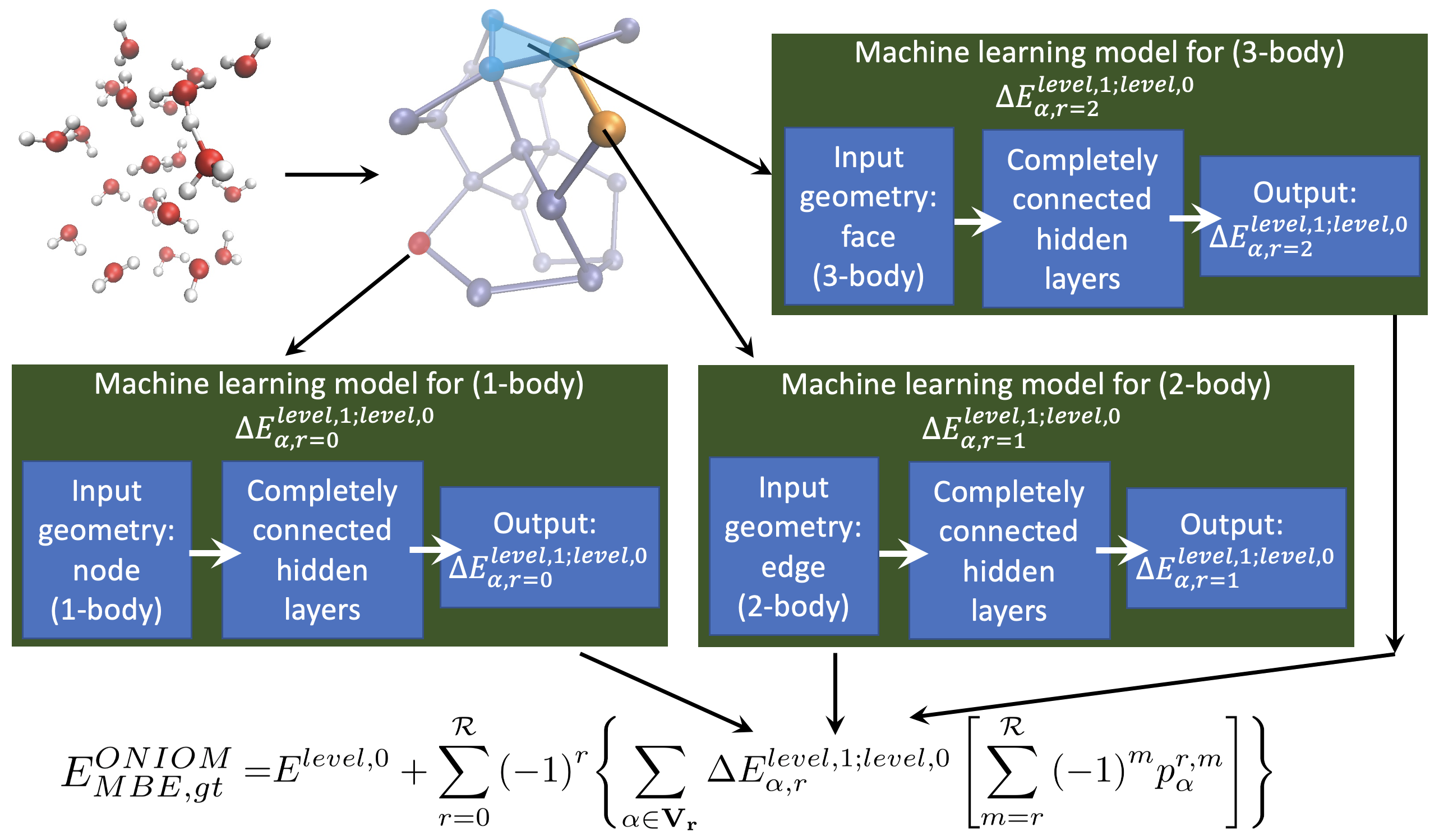 Summary: Over a series of publications we have introduced a graph-theoretic description for molecular fragmentation. Here, a system is divided into a set of nodes, or vertices, that are then connected through edges, faces, and higher-order simplexes to represent a collection of spatially overlapping and locally interacting subsystems. Each such subsystem is treated at two levels of electronic structure theory, and the result is used to construct many-body expansions that are then embedded within an ONIOM-scheme. These expansions converge rapidly with many-body order (or graphical rank) of subsystems and have been previously used for ab initio molecular dynamics (AIMD) calculations and for computing multidimensional potential energy surfaces. Specifically, in all these cases we have shown that CCSD and MP2 level AIMD trajectories and potential surfaces may be obtained at density functional theory cost. The approach has been demonstrated for gas-phase studies, for condensed phase electronic structure, and also for basis set extrapolation-based AIMD. Recently, this approach has also been used to derive new quantum-computing algorithms that enormously reduce the quantum circuit depth in a circuit-based computation of correlated electronic structure. In this publication, we introduce (a) a family of neural networks that act in parallel to represent, efficiently, the post-Hartree−Fock electronic structure energy contributions for all simplexes (fragments), and (b) a new k-means-based tessellation strategy to glean training data for high-dimensional molecular spaces and minimize the extent of training needed to construct this family of neural networks. The approach is particularly useful when coupled cluster accuracy is desired and when fragment sizes grow in order to capture nonlocal interactions accurately. The unique multidimensional k-means tessellation/clustering algorithm used to determine our training data for all fragments is shown to be extremely efficient and reduces the needed training to only 10% of data for all fragments to obtain accurate neural networks for each fragment. These fully connected dense neural networks are then used to extrapolate the potential energy surface for all molecular fragments, and these are then combined as per our graph-theoretic procedure to transfer the learning process to a full system energy for the entire AIMD trajectory at less than one-tenth the cost as compared to a regular fragmentation-based AIMD calculation.
Summary: Over a series of publications we have introduced a graph-theoretic description for molecular fragmentation. Here, a system is divided into a set of nodes, or vertices, that are then connected through edges, faces, and higher-order simplexes to represent a collection of spatially overlapping and locally interacting subsystems. Each such subsystem is treated at two levels of electronic structure theory, and the result is used to construct many-body expansions that are then embedded within an ONIOM-scheme. These expansions converge rapidly with many-body order (or graphical rank) of subsystems and have been previously used for ab initio molecular dynamics (AIMD) calculations and for computing multidimensional potential energy surfaces. Specifically, in all these cases we have shown that CCSD and MP2 level AIMD trajectories and potential surfaces may be obtained at density functional theory cost. The approach has been demonstrated for gas-phase studies, for condensed phase electronic structure, and also for basis set extrapolation-based AIMD. Recently, this approach has also been used to derive new quantum-computing algorithms that enormously reduce the quantum circuit depth in a circuit-based computation of correlated electronic structure. In this publication, we introduce (a) a family of neural networks that act in parallel to represent, efficiently, the post-Hartree−Fock electronic structure energy contributions for all simplexes (fragments), and (b) a new k-means-based tessellation strategy to glean training data for high-dimensional molecular spaces and minimize the extent of training needed to construct this family of neural networks. The approach is particularly useful when coupled cluster accuracy is desired and when fragment sizes grow in order to capture nonlocal interactions accurately. The unique multidimensional k-means tessellation/clustering algorithm used to determine our training data for all fragments is shown to be extremely efficient and reduces the needed training to only 10% of data for all fragments to obtain accurate neural networks for each fragment. These fully connected dense neural networks are then used to extrapolate the potential energy surface for all molecular fragments, and these are then combined as per our graph-theoretic procedure to transfer the learning process to a full system energy for the entire AIMD trajectory at less than one-tenth the cost as compared to a regular fragmentation-based AIMD calculation.
72 Graph-|Q⟩⟨C|, a Graph-Based Quantum/Classical Algorithm for Efficient Electronic Structure on Hybrid Quantum/Classical Hardware Systems: Improved Quantum Circuit Depth Performance
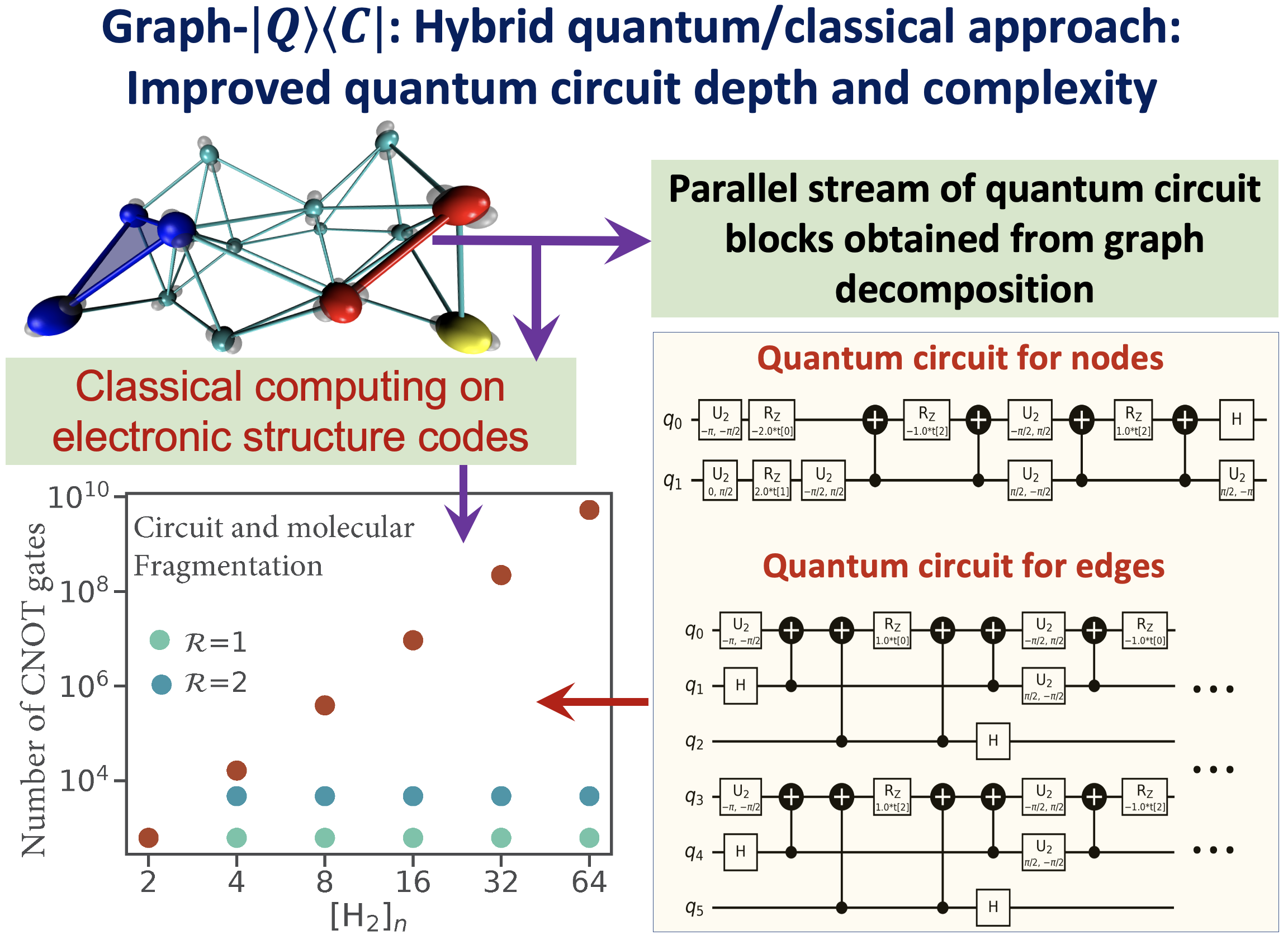 Juncheng Harry Zhang, and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 18, 2885 (2022).
Juncheng Harry Zhang, and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 18, 2885 (2022).
Summary: Recently, multiple quantum computing technologies have emerged as potential alternative computational platforms to address complex computational challenges. Additionally, algorithms to approximate electron correlation problems, for small molecular systems, and quantum nuclear dynamics problems have been implemented on quantum hardware devices. However, application of standard quantum circuit models to treat electronic structure problems leads to a rapid increase in the circuit depth and the number of quantum gates. This contributes greatly to the accumulated error during quantum propagation. This paper outlines a new hybrid quantum+classical algorithm based on a graph-theoretic approach to molecular fragmentation and is geared toward performing electron correlation calculations, potentially on an ensemble of quantum and classical hardware systems. The algorithm studied here is referred to as the “Graph-|Q⟩⟨C|” algorithm since it contains an independent set of classical and quantum algorithmic components inside a single umbrella. That is, the overall computational workload is partitioned, through graph theory based on computational complexity analysis, into (a) classical computing sections that are carried out on traditional classical electronic structure packages, and (b) quantum computing sections that are carried out using quantum circuit models. Furthermore, the Graph-|Q⟩⟨C| algorithm is quantum hardware-agnostic and is developed with the goal to be implemented on all quantum hardware technologies, and, in fact, is designed to be used on an ensemble of such quantum hardware systems for any given calculation. In essence, our Graph-|Q⟩⟨C| algorithm yields a new approach that reduces the required quantum circuit depth, the number of quantum gates, and the number of CNOT gates (by several orders of magnitude) that contribute to error accumulation, through a graph-theory-based projection operator formalism. Thus, given this reduction, our algorithm, potentially improves the quantum algorithmic efficiency, provides a new avenue for quantum resource management, and also reduces the accumulation of errors during the demonstrated electronic structure calculations on quantum hardware. Given the limitations of quantum circuit gate fidelities within the gate model, this algorithm, we expect, will become a central piece in the quantum/classical computing of chemical systems.
71 Mapping quantum chemical dynamics problems to spin-lattice simulators
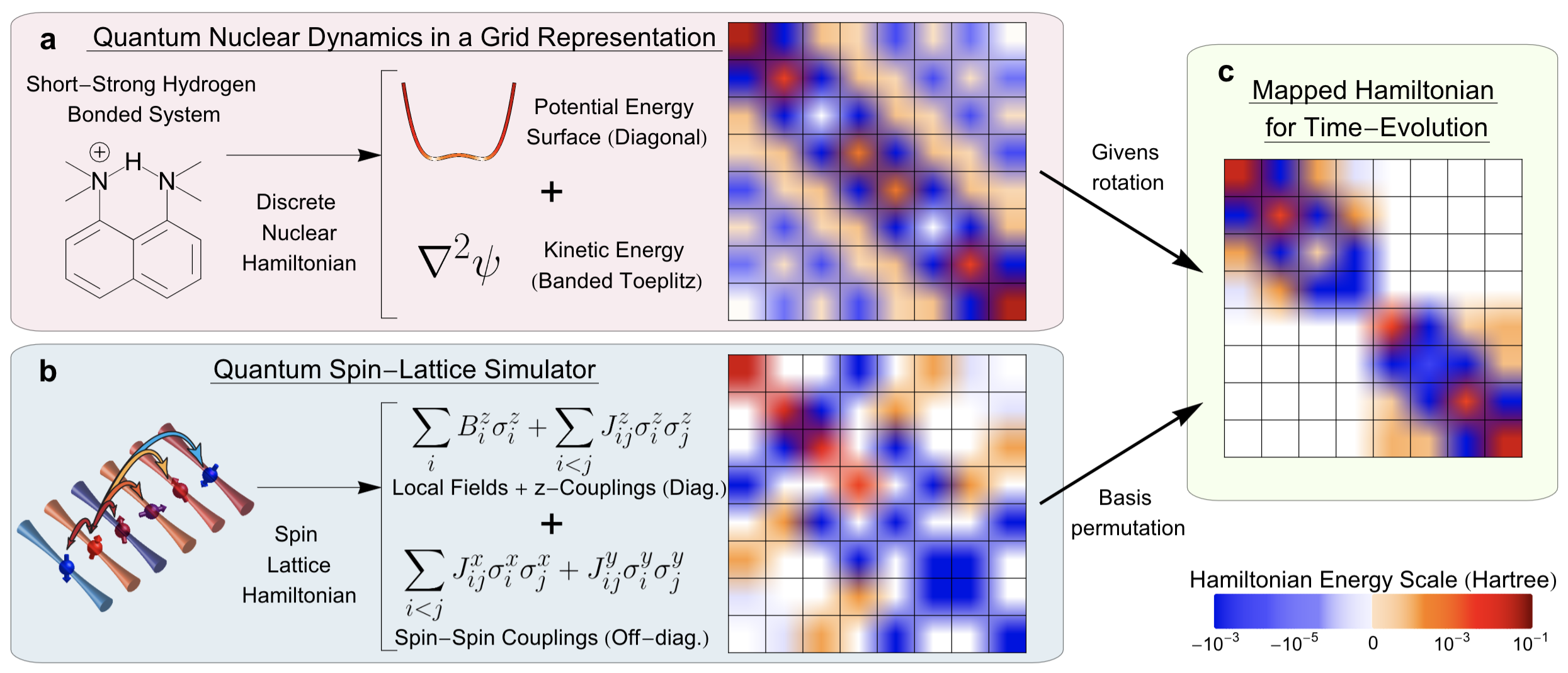 Debadrita Saha, Srinivasan S. Iyengar, Philip Richerme, Jeremy M. Smith, and Amr Sabry, J. Chem. Theory and Comput. 17, 6713 (2021).
Debadrita Saha, Srinivasan S. Iyengar, Philip Richerme, Jeremy M. Smith, and Amr Sabry, J. Chem. Theory and Comput. 17, 6713 (2021).
Summary: The accurate computational determination of chemical, materials, biological, and atmospheric properties has critical impact on a wide range of health and environmental problems, but is deeply limited by the computational scaling of quantum-mechanical methods. The complexity of quantum-chemical studies arises from the steep algebraic scaling of electron correlation methods, and the exponential scaling in studying nuclear dynamics and molecular flexibility. To date, efforts to apply quantum hardware to such quantum chemistry problems have focused primarily on electron correlation. Here, we provide a framework which allows for the solution of quantum chemical nuclear dynamics by mapping these to quantum spin-lattice simulators.
70 Graph-theory based molecular fragmentation for efficient and accurate potential surface calculations in multiple dimensions
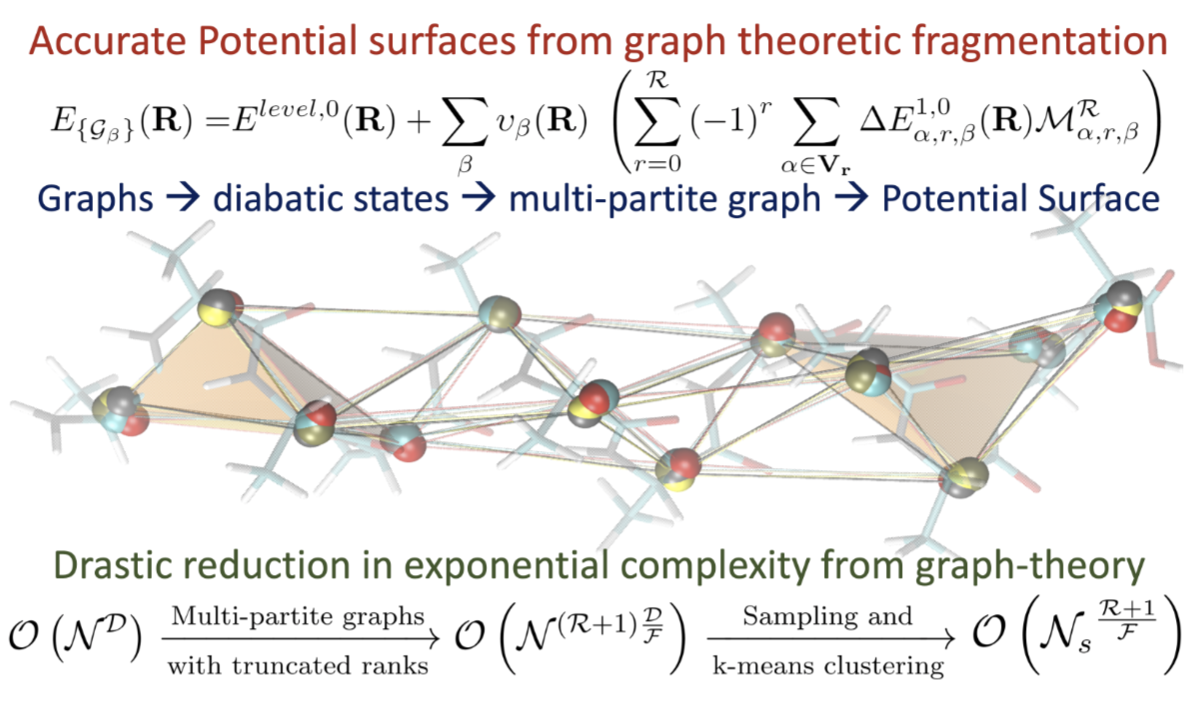 Anup Kumar, Nicole DeGregorio and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 17, 6671 (2021).
Anup Kumar, Nicole DeGregorio and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 17, 6671 (2021).
Summary: The accurate and efficient study of electronic structure and nuclear dynamics is at the core of multiple problems that are critical to materials, biological, and atmospheric research. However, these studies are deeply affected by the steep, polynomial scaling cost of electronic structure, and potentially exponential scaling of quantum nuclear dynamics. In this publication, we present a multi-topology molecular fragmentation approach, based on graph theory, to calculate multi-dimensional potential energy surfaces in agreement with post-Hartree-Fock levels of theory, but at DFT cost.
69 Weighted-Graph-Theoretic Methods for Many-Body Corrections within ONIOM: Smooth AIMD and the Role of High-Order ManyBody Terms
 Juncheng Harry Zhang, Timothy C. Ricard, Cody Haycraft, and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 17, 2672 (2021).
Juncheng Harry Zhang, Timothy C. Ricard, Cody Haycraft, and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 17, 2672 (2021).
Summary: We present a weighted-graph-theoretic approach to adaptively compute contributions from many-body approximations for smooth and accurate post-Hartree−Fock (pHF) ab initio molecular dynamics (AIMD) of highly fluxional chemical systems
68 An efficient and accurate approach to estimate hybrid functional and large basis set contributions to condensed phase systems and molecule-surface interactions
![]() T. C. Ricard and S. S. Iyengar, J. Chem. Theory and Comput. 16, 4790 (2020). (Supporting Information)
Summary: We present an efficient approach to perform accurate hybrid DFT electronic structure calculations for condensed phase systems, at pure DFT cost.
T. C. Ricard and S. S. Iyengar, J. Chem. Theory and Comput. 16, 4790 (2020). (Supporting Information)
Summary: We present an efficient approach to perform accurate hybrid DFT electronic structure calculations for condensed phase systems, at pure DFT cost.
67 Embedded, graph-theoretically defined many-body approximations for wavefunction-in-DFT and DFT-in-DFT: applications to gas- and condensed-phase AIMD, and potential surfaces for quantum nuclear effects
Timothy C. Ricard, Anup Kumar and Srinivasan S. Iyengar, Int. J. Quant. Chem. In Press. . Special issue on “Quantum Embedding Electronic Structure Methods”. 120, e26244 (2020). Summary: We discuss a graph theoretic approach to adaptively compute many-body-approximations in an efficient manner to perform (a) accurate post-Hartree-Fock AIMD at DFT cost for medium to large sized molecular clusters, (b) hybrid DFT electronic structure calculations for condensed phase simulations at the cost of pure density functionals, (c) reduced cost on-the-fly basis extrapolation for gas-phase AIMD and condensed phase studies, and (d) similar basis-set extrapolations for condensed phase calculations, and (e) accurate post-Hartree-Fock level potential energy surfaces at DFT cost for quantum nuclear effects.
66 Fragment-based electronic structure for potential energy surfaces using a superposition of fragmentation topologies
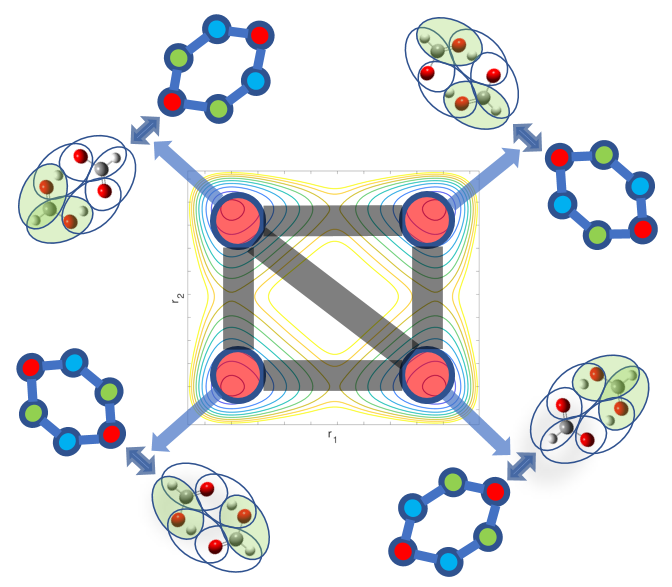 A. Kumar and S. S. Iyengar, "Fragment-based electronic structure for potential energy surfaces using a superposition of fragmentation topologies", J. Chem. Theory and Comput. 15, 5769 (2019).
A. Kumar and S. S. Iyengar, "Fragment-based electronic structure for potential energy surfaces using a superposition of fragmentation topologies", J. Chem. Theory and Comput. 15, 5769 (2019).
Summary: Molecular fragmentation methods developed by us and by several groups have made it possible to perform accurate electronic structure calculations in large systems. However, problems exist with these ideas when are applied to molecular potential surface calculations where fragments may change as the nuclei move. We develop a Fragment-Based electronic structure method which adaptively utilizes multiple graphical representations for a given molecular system and use these to compute smooth potential energy surfaces in an efficent manner. Graph theory based representations are used to efficiently compute and represent the local interactions between coarse grain units of molecules leading to post-Hartree-Fock accurate results at DFT cost. Benchmarks are provided on protonated waterwire systems ubiquitous in numerous biological ion channels and in material applications.
65 Challenges in constructing accurate methods for hydrogen transfer reactions in large biological assemblies: rare events sampling for mechanistic discovery and tensor networks for quantum nuclear effects
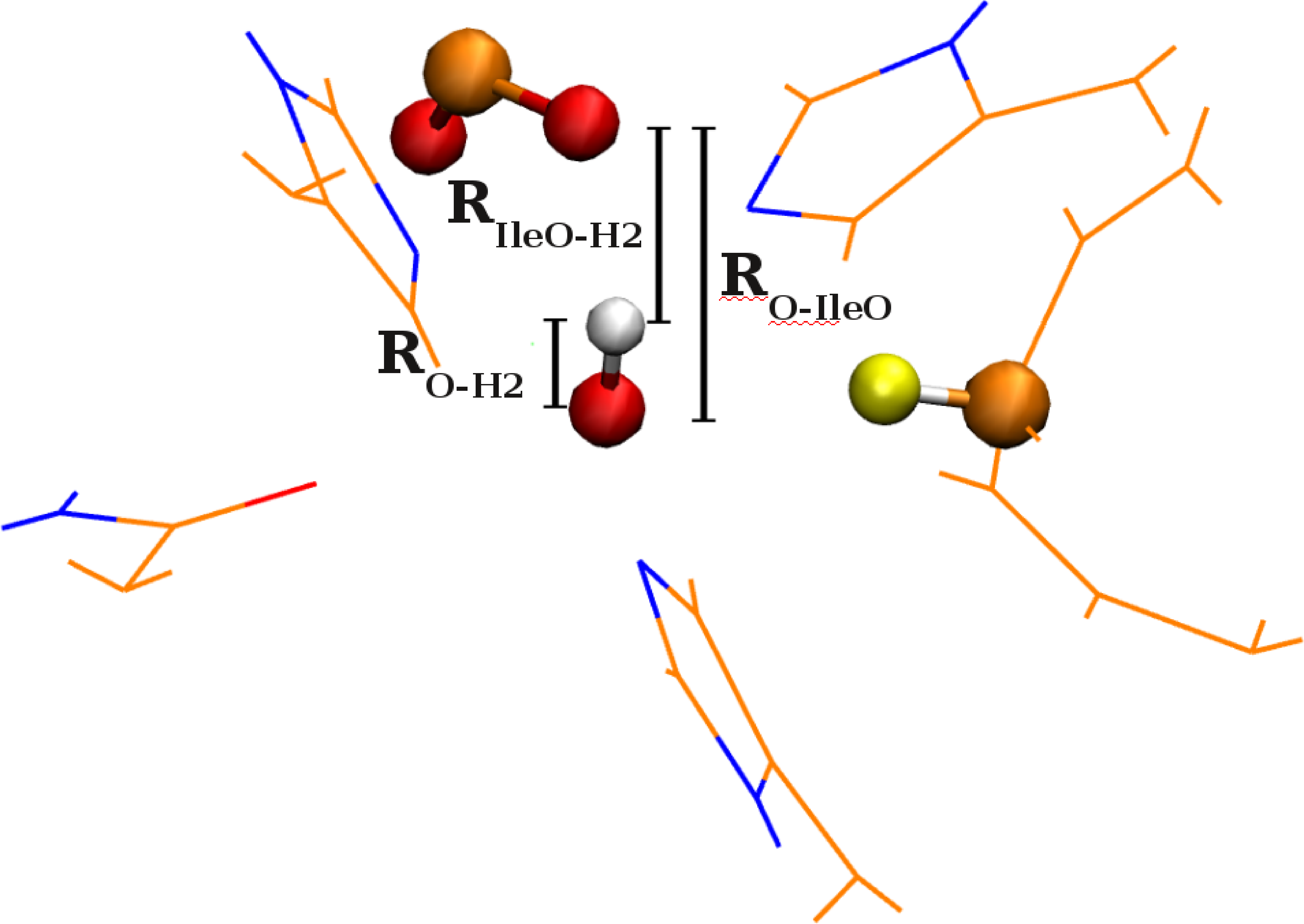 N. DeGregorio and S. S. Iyengar, "Challenges in constructing accurate methods for hydrogen transfer reactions in large biological assemblies: rare events sampling for mechanistic discovery and tensor networks for quantum nuclear effects", Faraday Discussions 221, 379-405 (2019).
N. DeGregorio and S. S. Iyengar, "Challenges in constructing accurate methods for hydrogen transfer reactions in large biological assemblies: rare events sampling for mechanistic discovery and tensor networks for quantum nuclear effects", Faraday Discussions 221, 379-405 (2019).
Summary: The computational complexities arising in hydrogen transfer reactions within the active site of the enzymes is addressed through a new formalism of rare-events sampling for ab initio dynamics where wuantum nuclear effects are included through tensor networks. We begin with an ab initio molecular dynamics adaptation of the Caldeira–Leggett system bath Hamiltonian and the direct application of this method helps discover the active participation of isoleucine-839 in facilitating the hydrogen transfer in SLO-1. This was initially noted in J. Phys. Chem. B. 119, 9532 (2015) and J. Phys. Chem. B 116 , 10145 (2012) (see below). The rare event sampled reaction pathways are then complemented through quantum nuclear dynamics on potential energy surfaces along the biased molecular dynamics trajectories to obtain critical insights on quantum nuclear effects. Improved representation of quantum dynamics data, such as potential surfaces and wavepacket evolutions, is obtained using tensor networks.
64 Adaptive Dimensional Decoupling for Compression of Quantum Nuclear Wave Functions and Efficient Potential Energy Surface Representations through Tensor Network Decomposition
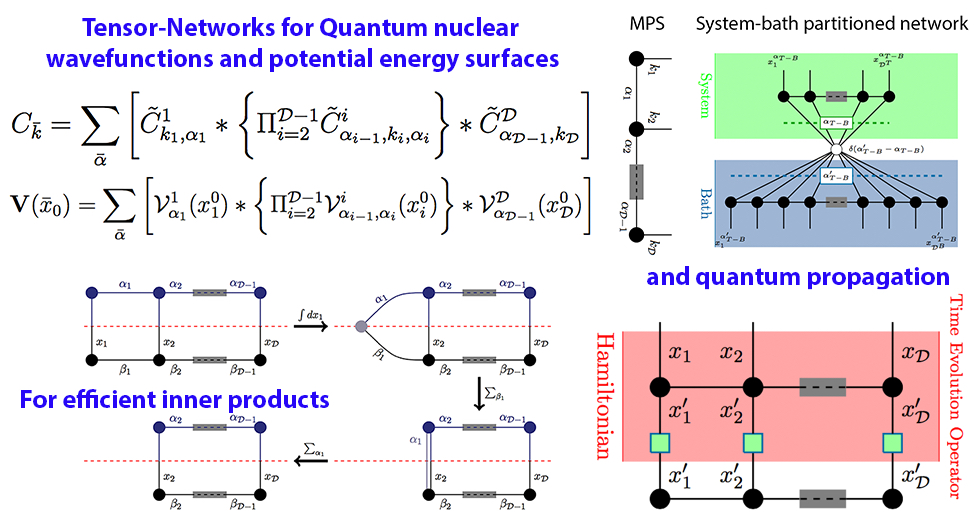 N. DeGregorio and S. S. Iyengar, "Adaptive Dimensional Decoupling for Compression of Quantum Nuclear Wave Functions and Efficient Potential Energy Surface Representations through Tensor Network Decomposition", J. Chem. Theory and Comput. 15, 2780 (2019).
N. DeGregorio and S. S. Iyengar, "Adaptive Dimensional Decoupling for Compression of Quantum Nuclear Wave Functions and Efficient Potential Energy Surface Representations through Tensor Network Decomposition", J. Chem. Theory and Comput. 15, 2780 (2019).
Summary: An approach to reduce the computational complexity and storage pertaining to quantum nuclear wave functions and potential energy surfaces is discussed. The method utilizes tensor networks; two specific forms of tensor networks are considered. (a) The well-known matrix product state is evaluated. (b) The wave function and potential energy surface space is initially partitioned into “system” and “bath” degrees of freedom, following which the individual system and bath tensors are in turn decomposed as matrix product states. Using these methods drastically reduces the overall storage and computational cost.
63 Photoelectrons are not always quite free
Jarrett L. Mason, Josey E. Topolski, Joshua Ewigleben, Srinivasan S. Iyengar, and Caroline Chick Jarrold, "Photoelectrons are not always quite free", J. Phys. Chem. Lett. 10, 144 (2019).
62 Efficiently capturing weak interactions in ab initio molecular dynamics with on the fly basis set extrapolation
T. C. Ricard and S. S. Iyengar, "Efficiently capturing weak interactions in ab initio molecular dynamics with on the fly basis set extrapolation", J. Chem. Theory and Comput. 14, 5535 (2018).
61 Exotic electronic structures in SmxCe3-xOy (x=0-3; y=2-4) clusters, and the effect of high neutral density of low-lying states on photodetachment transition intensities
J. E. Topolski, J. O. Kafader, V. Marrero-Colon, S. S. Iyengar H. P. Hratchian and C. C. Jarrold, "Exotic electronic structures in SmxCe3-xOy (x=0-3; y=2-4) clusters, and the effect of high neutral density of low-lying states on photodetachment transition intensities", J. Chem. Phys. 149, 054305 (2018).
60 Adaptive, geometric networks for efficient coarse-grained ab initio molecular dynamics with post-Hartree-Fock accuracy
T. C. Ricard, C. Haycraft, and S. S. Iyengar, "Adaptive, geometric networks for efficient coarse-grained ab initio molecular dynamics with post-Hartree-Fock accuracy", J. Chem. Theory and Comput. 14, 30 (2018).
59 Efficient and adaptive methods for computing accurate potential surfaces for quantum nuclear effects: Applications to hydrogen transfer reactions
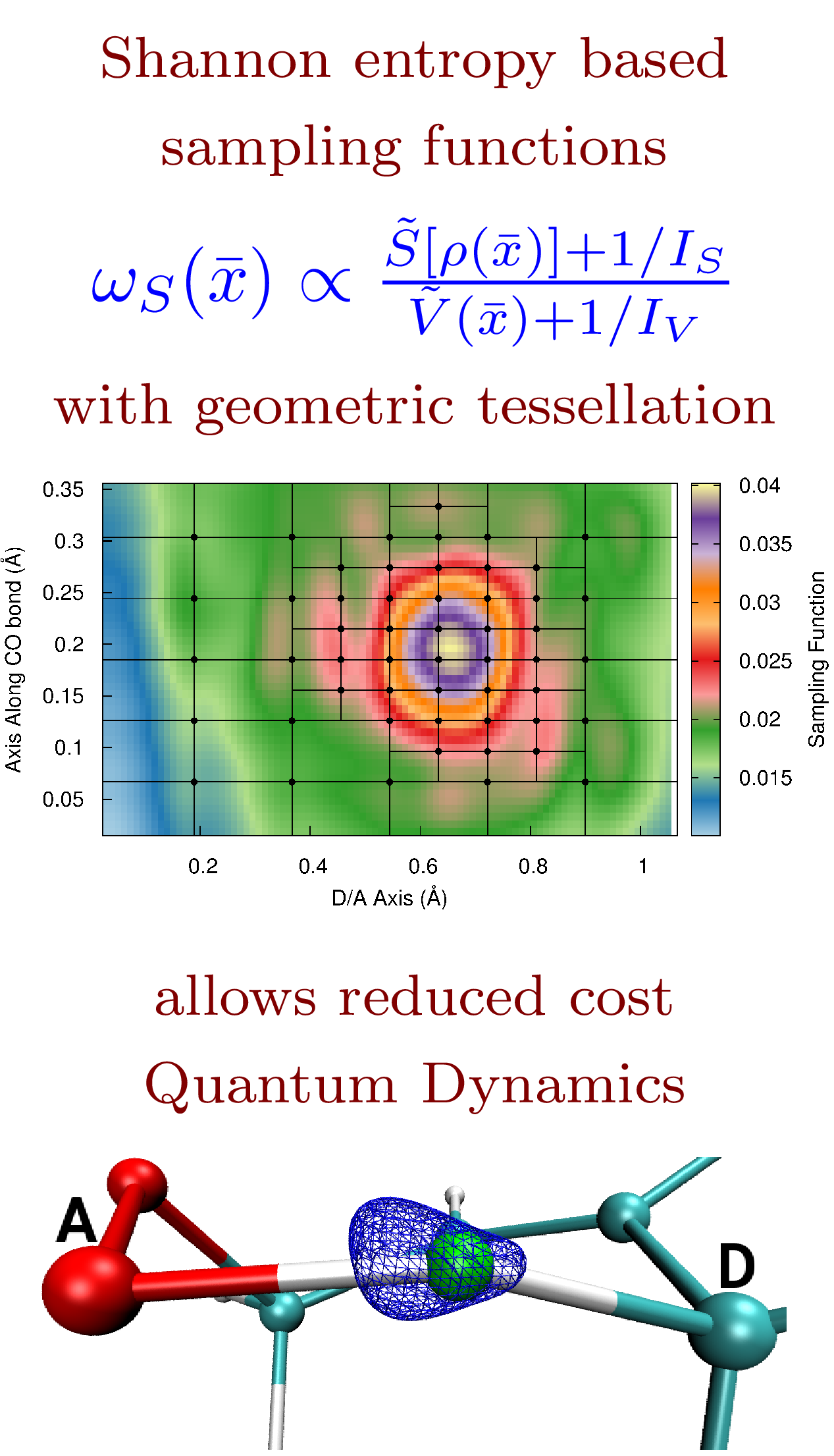 N. DeGregorio and S. S. Iyengar, "Efficient and adaptive methods for computing accurate potential surfaces for quantum nuclear effects: Applications to hydrogen transfer reactions" J. Chem. Theory and Comput. 14, 30 (2018).
N. DeGregorio and S. S. Iyengar, "Efficient and adaptive methods for computing accurate potential surfaces for quantum nuclear effects: Applications to hydrogen transfer reactions" J. Chem. Theory and Comput. 14, 30 (2018).
Summary: The exponentially scaling multi-dimensional nuclear potential energy surface space is tesselated using (a) Shannon information entropy, and (b) a sampling function determiend by the potential energy, its nuclear gradients and wavepacket density. The methods are used to efficiently compute potential surfaces for hydrogen transfer problems of interest in atmospheric and biological problems.
58 Proton relays in anomalous carbocations dictate spectroscopy, stability and mechanisms: Case studies on C2H5+ and C3H3+
L. M. Sager and S. S. Iyengar, "Proton relays in anomalous carbocations dictate spectroscopy, stability and mechanisms: Case studies on C2H5+ and C3H3+" Phys. Chem. Chem. Phys. 19, 27801 (2017).
57 The electron shuttle: Cerium influences samaraium 4f orbital occupancy in heteronuclear Ce-Sm oxide clusters
J. O. Kafader, J. E. Topolski, V. Marrero-Colon, S. S. Iyengar and C. C. Jarrold, "The electron shuttle: Cerium influences samaraium 4f orbital occupancy in heteronuclear Ce-Sm oxide clusters". J. Chem. Phys. 146, 194310 (2017).
56 Efficient, 'On-the-fly', Born-Oppenheimer and Car-Parrinello-type dynamics with coupled cluster (CCSD) accuracy through fragment based electronic structure
C. Haycraft, J. Li and S. S. Iyengar, "Efficient, 'On-the-fly', Born-Oppenheimer and Car-Parrinello-type dynamics with coupled cluster (CCSD) accuracy through fragment based electronic structure" J. Chem. Theory and Comput. 13, 1885 (2017). Supporting Information: Click here
55 A Grotthuss-like proton shuttle in the anomalous C2H3+ Carbocation: Energetic and vibrational properties for isotopologues
J. Li, A. B. Pacheco, K. Raghavachari and S. S. Iyengar, "A Grotthuss-like proton shuttle in the anomalous C2H3+ Carbocation: Energetic and vibrational properties for isotopologues" Phys. Chem. Chem. Phys. 18, 29395 (2016). (Click here for supporting information.)
54 Hybrid extended Lagrangian, post-Hartree-Fock Born-Oppenheimer ab initio molecular dynamics with fragment-based electronic structure
J. Li, C. Haycraft and S. S. Iyengar "Hybrid extended Lagrangian, post-Hartree-Fock Born-Oppenheimer ab initio molecular dynamics with fragment-based electronic structure". J. Chem. Theory and Comput. 12, 2493 (2016).
53 Ab initio molecular dynamics using recursive, spatially separated, overlapping model subsystems mixed within an ONIOM based fragmentation energy extrapolation technique
J. Li and S. S. Iyengar "Ab initio molecular dynamics using recursive, spatially separated, overlapping model subsystems mixed within an ONIOM based fragmentation energy extrapolation technique". J. Chem. Theory and Comput. 11, 3978 (2015).
52 Active site dynamical effects that affect the hydrogen transfer rate-limiting step in the catalysis of linoleic acid by soybean lipoxygenase-1 (SLO-1): Primary and Secondary Isotope effects
P. Phatak, J. Vanderley, J. deBrota, J. Li and S. S. Iyengar, "Active site dynamical effects that affect the hydrogen transfer rate-limiting step in the catalysis of linoleic acid by soybean lipoxygenase-1 (SLO-1): Primary and Secondary Isotope effects". J. Phys. Chem. B. 119, 9532 (2015).
51 A multi-wavelet treatment of the quantum subsystem in quantum wavepacket ab-initio molecular dynamics through an hierarchical partitioning of momentum space
A. H. Prociuk and S. S. Iyengar, "A multi-wavelet treatment of the quantum subsystem in quantum wavepacket ab-initio molecular dynamics through an hierarchical partitioning of momentum space", J. Chem. Theory Comput. 10, 2950 (2014).
50 Multi-configurational generalizations to the "on-the'fly" electronic structure calculations within quantum wavepacket ab initio molecular dynamics (QWAIMD): Applications to the study of vibrational properties in hydrogen bonded systems
J. Li, X. Li and S. S. Iyengar, "Multi-configurational generalizations to the "on-the'fly" electronic structure calculations within quantum wavepacket ab initio molecular dynamics (QWAIMD): Applications to the study of vibrational properties in hydrogen bonded systems", J. Chem. Theory Comput. 10, 2265 (2014).
49 Teaching Thermodynamics and Kinetics to Advanced General Chemistry Students and to Upper-Level Undergraduate Students Using PV Diagrams
S. S. Iyengar and R. T. deSouza, "Teaching Thermodynamics and Kinetics to Advanced General Chemistry Students and to Upper-Level Undergraduate Students Using PV Diagrams", J. Chem. Ed. 91, 74 (2014).
48 Using Quantum Mechanics To Facilitate the Introduction of a Broad Range of Chemical Concepts to First-Year Undergraduate Students
R. T. deSouza and S. S. Iyengar, "Using Quantum Mechanics To Facilitate the Introduction of a Broad Range of Chemical Concepts to First-Year Undergraduate Students", J. Chem. Ed. 90, 717 (2013).
47 Constructing periodic phase space orbits from ab initio molecular dynamics trajectories to analyze vibrational spectra: case study of the Zundel (H5O2+) cation
S. M. Dietrick and S. S. Iyengar, "Constructing periodic phase space orbits from ab initio molecular dynamics trajectories to analyze vibrational spectra: case study of the Zundel (H5O2+) cation", In Press. J. Chem. Theory and Comp. 8 , 4876 (2012).
46 Gauging the flexibility of the active site in soybean lipoxygenase-1 (SLO-1) through an atom-centered density matrix propagation (ADMP) treatment that facilitates the sampling of rare events
P. Phatak, I. Sumner and S. S. Iyengar, "Gauging the flexibility of the active site in soybean lipoxygenase-1 (SLO-1) through an atom-centered density matrix propagation (ADMP) treatment that facilitates the sampling of rare events", J. Phys. Chem. B 116 , 10145 (2012).
45 'Pump-probe' atom-centered density matrix propagation studies to gauge anharmonicity and energy repartitioning in atmospheric reactive adducts: Case study of the OH + Isoprene and OH + Butadiene reaction intermediates
A. B. Pacheco, S. M. Dietrick, P. S. Stevens and S. S. Iyengar, "'Pump-probe' atom-centered density matrix propagation studies to gauge anharmonicity and energy repartitioning in atmospheric reactive adducts: Case study of the OH + Isoprene and OH + Butadiene reaction intermediates", J. Phys. Chem. A 116 , 4108 (2012).
44 The influence of water on anharmonicity, stability and vibrational energy distribution of hydrogen-bonded adducts in atmospheric reactions: Case study of Hydroxy Isoprene using ab-initio Molecular Dynamics
S. M. Dietrick, A. B. Pacheco, P. Phatak, P. S. Stevens and S. S. Iyengar, "The influence of water on anharmonicity, stability and vibrational energy distribution of hydrogen-bonded adducts in atmospheric reactions: Case study of Hydroxy Isoprene using ab-initio Molecular Dynamics", J. Phys. Chem. A 116 , 399 (2012).
43 Quantum Wave-packet Ab Initio Molecular dynamics for extended systems
X. Li and S. S. Iyengar, "Quantum Wave-packet Ab Initio Molecular dynamics for extended systems" J. Phys. Chem. A 115 , 6269 (2011).
42 Multi-Stage Ab-initio Quantum Wavepacket Dynamics for Electronic Structure and Dynamics in open systems: Momentum Representation, Coupled Electron Nuclear dynamics and External Fields
A. B. Pacheco and S. S. Iyengar, "Multi-Stage Ab-initio Quantum Wavepacket Dynamics for Electronic Structure and Dynamics in open systems: Momentum Representation, Coupled Electron Nuclear dynamics and External Fields" J. Chem. Phys. 134 , 074107 (2011).
41 Shannon information entropy based time-dependent deterministic sampling techniques for efficient on-the-fly quantum dynamics and electronic structure
D. Hocker, X. Li and S. S. Iyengar, "Shannon information entropy based time-dependent deterministic sampling techniques for efficient on-the-fly quantum dynamics and electronic structure" J. Chem. Theory and Comp. 7, 256 (2011).
40 Quantum wavepacket ab initio molecular dynamics: Generalizations using an extended Lagrangian treatment of diabatic states coupled through multi-reference electronic structure
X. Li and S. S. Iyengar, "Quantum wavepacket ab initio molecular dynamics: Generalizations using an extended Lagrangian treatment of diabatic states coupled through multi-reference electronic structure" J. Chem. Phys. 133, 184105 (2010).
39 A Multi-Stage Ab-initio Quantum Wavepacket Dynamics Formalism for Electronic Structure and Dynamics in Open Systems
A. B. Pacheco and S. S. Iyengar, "A Multi-Stage Ab-initio Quantum Wavepacket Dynamics Formalism for Electronic Structure and Dynamics in Open Systems" J. Chem. Phys. 133, 044105 (2010).
38 Isotope dependent, temperature regulated, energy repartitioning in a low-barrier, short-strong hydrogen bonded cluster
X. Li, J. Oomens, J. R. Eyler, D. T. Moore, S. S. Iyengar, "Isotope dependent, temperature regulated, energy repartitioning in a low-barrier, short-strong hydrogen bonded cluster" J. Chem. Phys. 132, 244301 (2010).
37 Analysis of Hydrogen Tunneling in an Enzyme Active Site Using von Neumann Measurements
I. Sumner and S. S. Iyengar, "Analysis of Hydrogen Tunneling in an Enzyme Active Site Using von Neumann Measurements" Journal of Chemical Theory and Computation 6 , 1698 (2010).
36 Computing vibrational properties in hydrogen bonded systems using Quantum wavepacket ab initio molecular dynamics
S. S. Iyengar, "Computing vibrational properties in hydrogen bonded systems using Quantum wavepacket ab initio molecular dynamics" Int. J. Quant. Chem. 109 , 3798 (2009).
35 Combining quantum wavepacket ab initio molecular dynamics (QWAIMD) with QM/MM and QM/QM techniques: Implementation blending ONIOM and empirical valence bond theory
I. Sumner and S. S. Iyengar, "Combining quantum wavepacket ab initio molecular dynamics (QWAIMD) with QM/MM and QM/QM techniques: Implementation blending ONIOM and empirical valence bond theory" J. Chem. Phys. 129, 054109 (2008).
34 Experimental and theoretical study of the kinetics of the OH + 1,3-butadiene reaction between 263 and 423 K at low pressure
D. Vimal, A. B. Pacheco, S. S. Iyengar and P. S. Stevens, "Experimental and theoretical study of the kinetics of the OH + 1,3-butadiene reaction between 263 and 423 K at low pressure". J. Phys. Chem. A 112, 7227 (2008).
33 Hydrogen tunneling in an enzyme active site: a quantum wavepacket dynamical perspective
S. S. Iyengar, I. Sumner and J. Jakowski "Hydrogen tunneling in an enzyme active site: a quantum wavepacket dynamical perspective". J. Phys. Chem. B 112, 7601 (2008).
32 Insights from first principles molecular dynamics studies towards infra-red multiple-photon and single-photon action spectroscopy: Case study of the proton-bound di-methyl ether dimer
X. Li, D. T. Moore and S. S. Iyengar, "Insights from first principles molecular dynamics studies towards infra-red multiple-photon and single-photon action spectroscopy: Case study of the proton-bound di-methyl ether dimer". J. Chem. Phys. 128 , 184308 (2008).
31 The study of dynamically averaged vibrational spectroscopy of atmospherically relevant clusters using ab initio molecular dynamics in conjunction with quantum wavepackets
S. S. Iyengar, X. Li and I. Sumner "The study of dynamically averaged vibrational spectroscopy of atmospherically relevant clusters using ab initio molecular dynamics in conjunction with quantum wavepackets". Adv. Quant. Chem. 55 , 333 (2008).
30 Multiscale Theory of Collective and Quasi-Particle Modes in Quantum Nanosystems
P. Ortoleva and S. S. Iyengar, "Multiscale Theory of Collective and Quasi-Particle Modes in Quantum Nanosystems". J. Chem. Phys 128 , 164716 (2008)
29 Quantum Wavepacket Ab Initio Molecular Dynamics: An approach for computing dynamically averaged vibrational spectra including critical nuclear quantum effects
I. Sumner and S. S. Iyengar "Quantum Wavepacket Ab Initio Molecular Dynamics: An approach for computing dynamically averaged vibrational spectra including critical nuclear quantum effects". J. Phys. Chem. A, 111 , 10313-10324 (2007).
28 Further analysis of the dynamically averaged vibrational spectrum for the 'magic' protonated 21-water cluster
S. S. Iyengar "Further analysis of the dynamically averaged vibrational spectrum for the 'magic' protonated 21-water cluster". J. Chem. Phys. 126 , 216101 (2007).
27 Can the four-coordinated, penta-valent oxygen in hydroxide water clusters be detected through experimental vibrational spectroscopy?
X. Li, V. E. Teige and S. S. Iyengar "Can the four-coordinated, penta-valent oxygen in hydroxide water clusters be detected through experimental vibrational spectroscopy?". J. Phys. Chem. A 111 , 4815-4820 (2007).
26 Computational Improvements to Quantum Wavepacket Ab Initio Molecular Dynamics using a potential-adapted, time-dependent deterministic sampling technique
J. Jakowski, I. Sumner and S. S. Iyengar, "Computational Improvements to Quantum Wavepacket Ab Initio Molecular Dynamics using a potential-adapted, time-dependent deterministic sampling technique". Journal of Chemical Theory and Computation 2 1203-1219 (2006).
25 Ab Initio Dynamics with wave-packets and density matrices
S. S. Iyengar, "Ab Initio Dynamics with wave-packets and density matrices". Theo. Chem. Accts. Special issue on "New Perspectives in Theoretical Chemistry" Vol. 116, 326-337 (2006)
24 The Properties of Ion-Water Clusters. II. Solvation structures of Na+, Cl- and H+ clusters as a function of temperature
C. J. Burnham, M. K. Petersen, T. J. F. Day, S. S. Iyengar and G. A. Voth, "The Properties of Ion-Water Clusters. II. Solvation structures of Na+, Cl- and H+ clusters as a function of temperature". J. Chem. Phys. 124 , 024327 (2006)
23 Quantum Wavepacket Ab Initio Molecular Dynamics: An approach to study quantum dynamics in large systems
S. S. Iyengar and J. Jakowski, "Quantum Wavepacket Ab Initio Molecular Dynamics: An approach to study quantum dynamics in large systems". J. Chem. Phys. 122 114105-114115 (2005).
22 Dynamical effects on vibrational and electronic spectra of HO2 water clusters: an ab initio molecular dynamics study
S. S. Iyengar, "Dynamical effects on vibrational and electronic spectra of HO2 water clusters: an ab initio molecular dynamics study". J. Chem. Phys. 123 , 084310 (2005)
21 The Properties of Ion-Water Clusters. I. The Protonated 21-Water Cluster
S. S. Iyengar, M. K. Petersen, C. J. Burnham, T. J. F. Day, V. E. Teige and G. A. Voth, "The Properties of Ion-Water Clusters. I. The Protonated 21-Water Cluster". J. Chem. Phys. 123 , 084309 (2005)
20 On the Amphiphilic Behavior of the Hydrated Proton: An Ab Initio Molecular Dynamics Study
S. S. Iyengar, T. J. F. Day, and G. A. Voth, "On the Amphiphilic Behavior of the Hydrated Proton: An Ab Initio Molecular Dynamics Study". Int. J. Mass Spectrometry 241, 197-204 (2005).
19 Comment on ``Curvy-steps approach to constraint-free extended-Lagrangian ab initio molecular dynamics, using atom-centered basis functions: Convergence toward Born-Oppenheimer trajectories
S. S. Iyengar, H. B. Schlegel, G. E. Scuseria, J. M. Millam and M. J. Frisch, "Comment on ``Curvy-steps approach to constraint-free extended-Lagrangian ab initio molecular dynamics, using atom-centered basis functions: Convergence toward Born-Oppenheimer trajectories''[J. Chem. Phys. 121, 11542 (2004)]". J. Chem. Phys. 123 , 027101 (2005)
18 The Hydrated Proton at the Water Liquid/Vapor Interface
M. K. Petersen, S. S. Iyengar, C. J. Burnham, T. J. F. Day, and G. A. Voth, "The Hydrated Proton at the Water Liquid/Vapor Interface". J. Phys. Chem. B 108 , 14804 (2004).
17 Effect of time-dependent basis functions and their superposition error on Atom-centered Density Matrix Propagation (ADMP): Connections to Wavelet theory of multi-resolution analysis
S. S. Iyengar and M. J. Frisch, "Effect of time-dependent basis functions and their superposition error on Atom-centered Density Matrix Propagation (ADMP): Connections to Wavelet theory of multi-resolution analysis". J. Chem. Phys.121, 5061 (2004)
16 Hybrid Ab-Initio/Empirical Molecular Dynamics: Combining the ONIOM Scheme with the Atom-centered Density Matrix Propagation (ADMP) Approach
N. Rega, S. S. Iyengar, G. A. Voth, H. B. Schlegel, T. Vreven and M. J. Frisch, "Hybrid Ab-Initio/Empirical Molecular Dynamics: Combining the ONIOM Scheme with the Atom-centered Density Matrix Propagation (ADMP) Approach". J. Phys. Chem. B 108 4210-4220 (2004).
15 Modeling Condensed-phase chemistry through molecular dynamics simulation
S. S. Iyengar, C. J. Burnham, M. K. Petersen, and G. A. Voth, "Modeling Condensed-phase chemistry through molecular dynamics simulation", Computers in Science and Engineering 5 Iss. 3, 31-35 (2003). (special issue on Computational Chemistry)
14 Atom-centered Density Matrix Propagation: Generalizations using Bohmian mechanics
S. S. Iyengar, H. B. Schlegel, and G. A. Voth, "Atom-centered Density Matrix Propagation: Generalizations using Bohmian mechanics", J. Phys. Chem. A. 107 7269-7277 (2003). (Festschrift in honor of Professor Donald J. Kouri)
13 Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian orbitals. IV. Formal Analysis of the deviations from Born-Oppenheimer dynamics
S. S. Iyengar, H. B. Schlegel, G. A. Voth, J. M. Millam, G. E. Scuseria, M. J. Frisch, "Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian orbitals. IV. Formal Analysis of the deviations from Born-Oppenheimer dynamics". Israel J. Chem 7 191, 2002. (Special Issue on Condensed Phase Dynamics)
12 Quantum reactive scattering in three dimensions using hyperspherical (APH) coordinates: Periodic Distributed Approximating Functionals (PDAF) method for surface functions
K. Zhang, G. A. Parker, D. J. Kouri, D. K. Hoffman, S. S. Iyengar, "Quantum reactive scattering in three dimensions using hyperspherical (APH) coordinates: Periodic Distributed Approximating Functionals (PDAF) method for surface functions". J. Chem. Phys.118, 569 (2003).
11 Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian orbitals. III. Comparison with Born-Oppenheimer Dynamics
H. B. Schlegel, S. S. Iyengar, X. Li, J. M. Millam, G. A. Voth, G. E. Scuseria, M. J. Frisch, "Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian orbitals. III. Comparison with Born-Oppenheimer Dynamics".J. Chem. Phys. 117, 8694 (2002).
10 Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian orbitals. II. Generalizations based on Mass-weighting, Idempotency, Energy Conservation and Choice of Initial Conditions
S. S. Iyengar, H. B. Schlegel, J. M. Millam, G. A. Voth, G. E. Scuseria, M. J. Frisch, "Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian orbitals. II. Generalizations based on Mass-weighting, Idempotency, Energy Conservation and Choice of Initial Conditions" J. Chem. Phys. 115, 10291 (2001).
9 The Challenge of Creating Accurate and Effective Kinetic Energy Functionals
S. S. Iyengar, M. Ernzerhoff, S.N. Maximoff, G. E. Scuseria, "The Challenge of Creating Accurate and Effective Kinetic Energy Functionals". Phys. Rev. A 63 (2001) 052508
8 Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian Orbitals
H. B. Schlegel, J. M. Millam, S. S. Iyengar, G. A. Voth, A. D. Daniels, G. E. Scuseria, M. J. Frisch, "Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian Orbitals". J. Chem. Phys. 114, 9758 (2001).
7 Particular and Homogeneous Solutions of Time-Independent Wavepacket Schrodinger Equations: Calculations Using a Subset of Eigenstates of Undamped or Damped Hamiltonians
S. S. Iyengar, D. J. Kouri, D. K. Hoffman, "Particular and Homogeneous Solutions of Time-Independent Wavepacket Schrodinger Equations: Calculations Using a Subset of Eigenstates of Undamped or Damped Hamiltonians". Theoretical Chemistry Accounts. 104 (2000) 471-483.
6 Bounding the Extrapolated Correlation Energy Using Pade Approximants
S. S. Iyengar, G. E. Scuseria, A. Savin, "Bounding the Extrapolated Correlation Energy Using Pade Approximants". Int. J. Quant. Chem. 79 (2000) 222-234
5 Estimating the Upper and Lower Bounds for the Eigenvalues of any matrix
S. S. Iyengar, D. J. Kouri, G. A. Parker, D. K. Hoffman, "Estimating the Upper and Lower Bounds for the Eigenvalues of any matrix". Theoretical Chemistry Accounts. 103 (2000) 507-517.
4 Symmetry-Adapted Distributed Approximating Functionals: Theory and application to the ro-vibrational states of H3+
S. S. Iyengar, G. A. Parker, D. J. Kouri, D. K. Hoffman, "Symmetry-Adapted Distributed Approximating Functionals: Theory and application to the ro-vibrational states of H3+". J. Chem. Phys. 110, 10283 (1999).
3 Further Analysis of Solutions to the Time-Independent Wavepacket Equations of Quantum Dynamics II: Scattering as a Continuous Function of Energy Using Finite, Discrete Approximate Hamiltonians
Y. Huang, S. S. Iyengar, D. J. Kouri, D. K. Hoffman, "Further Analysis of Solutions to the Time-Independent Wavepacket Equations of Quantum Dynamics II: Scattering as a Continuous Function of Energy Using Finite, Discrete Approximate Hamiltonians" J. Chem. Phys. 105, 927 (1996).
2 Inclusion of molecular flexibility in Partition Coefficient calculations on oligopeptides using stochastic sampling
N. G. J. Richards, P. B. Williams, S. S. Iyengar, Proceedings of the twenty-seventh Hawaii International Conference on System Sciences 203 (1994).
1 Semi-empirical calculations on the mechanism of enniatin B mediated transport of sodium ions
S. N. Datta, S. S. Iyengar, "Semi-empirical calculations on the mechanism of enniatin B mediated transport of sodium ions" Chem. Phys. Lett. 183, 491 (1991).
 The College of Arts
The College of Arts