

Graph theoretic methods for dynamics of electrons and nuclei with post-Hartree-Fock accuracy
The accurate treatment of molecular properties often requires the correlated study of electronic structure with extensive basis sets. But the size of systems that can be considered by standard approaches in electronic structure is greatly limited due to the intrinsic (steeply algebraic) computational scaling of electron correlation methods and the number and quality of basis functions used. Although there has been significant progress through algorithmic improvements for the treatment of electron correlation, the computational expense is still often too high for most systems of chemical interest. This is especially true when dynamics calculations are to be performed with on-the-fly electronic structure or when quantum nuclear effects are to be studied.
![]()
In a series of publications (see below) we have shown how graph-theoretic methods may be used to adaptively construct many-body approximations to potential surfaces and AIMD calculations. In the process both extended Lagrangian as well as Born-Oppenheimer based ab initio molecular dynamics simulations can be performed at accuracy comparable to CCSD and MP2 levels of theory with DFT-computational cost. Hence for the studies presented below, we presented Car-Parrinello-style dynamics, but with CCSD accuracy. Similarly, we have shown how multiple graphical representations of molecular systems may be simultaneously utilized to construct accurate potential surfaces in agreement with MP2 and CCSD levels of theory, again at DFT cost.
Furthermore we have also shown how condensed-phase simulations on interfaces and liquids may be constructed with hybrid DFT accuracy at gradient-corrected DFT accuracy. The approach utilizes graph-theoretic methods.
Representative publications
82 A reformulation of all ONIOM-type molecular fragmentation approaches using graph theory-based projection operators: Applications to dynamics, molecular potential surfaces, and machine learning and quantum computing
S. S. Iyengar, Timothy C. Ricard, Xiao Zhu, “A reformulation of all ONIOM-type molecular fragmentation approaches using graph theory-based projection operators: Applications to dynamics, molecular potential surfaces, and machine learning and quantum computing” J. Phys. Chem. A, ASAP (2023). J. Phys. Chem. A 128, 466 (2024).
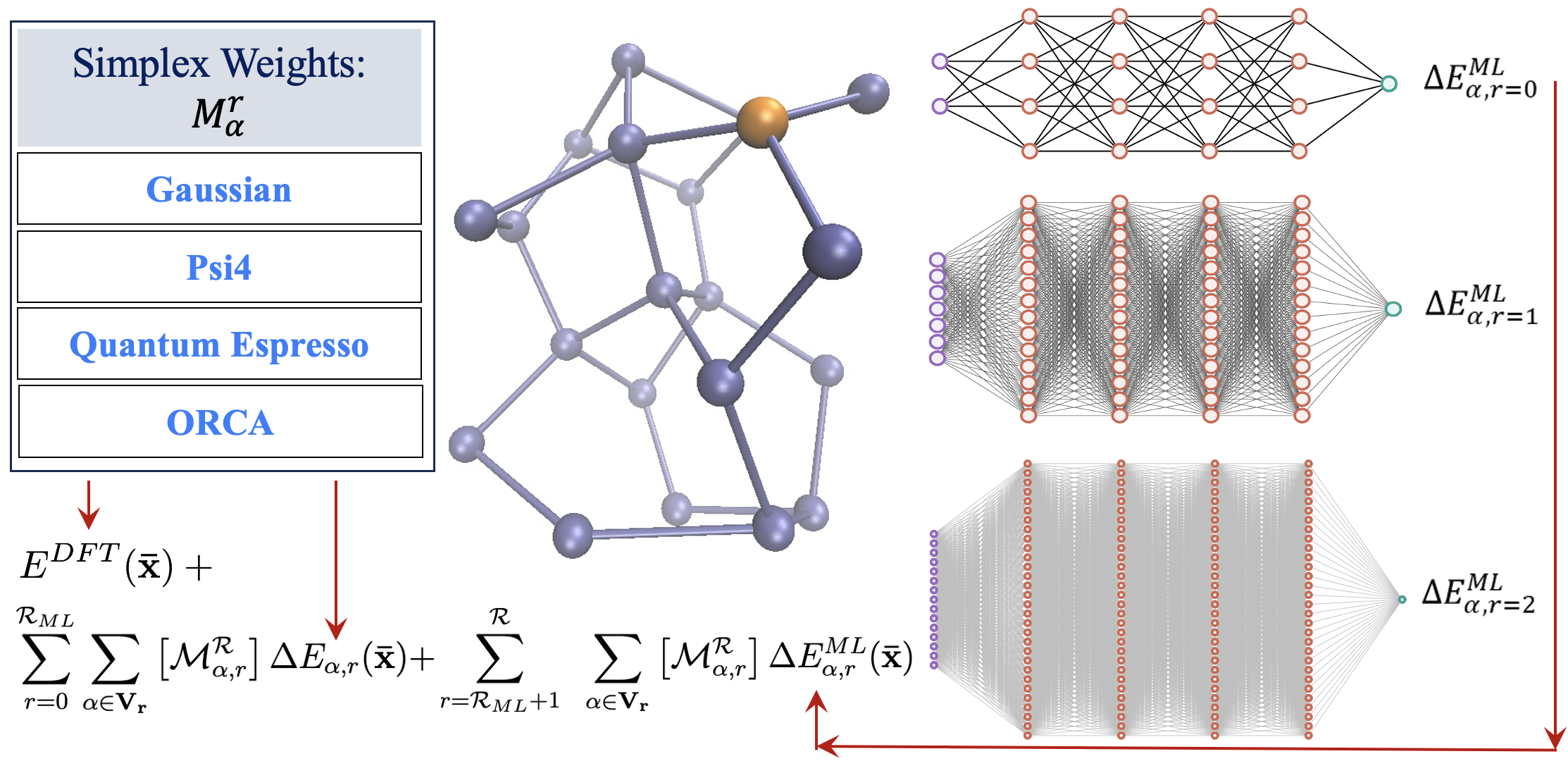 Summary: We present a graph-theory-based reformulation of all ONIOM-based molecular fragmentation methods. We discuss applications to (a) accurate post-Hartree–Fock AIMD that can be conducted at DFT cost for medium-sized systems, (b) hybrid DFT condensed-phase studies at the cost of pure density functionals, (c) reduced cost on-the-fly large basis gas-phase AIMD and condensed-phase studies, (d) post-Hartree–Fock-level potential surfaces at DFT cost to obtain quantum nuclear effects, and (e) novel transfer machine learning protocols derived from these measures. Additionally, in previous work, the unifying strategy discussed here has been used to construct new quantum computing algorithms. Thus, we conclude that this reformulation is robust and accurate.
Summary: We present a graph-theory-based reformulation of all ONIOM-based molecular fragmentation methods. We discuss applications to (a) accurate post-Hartree–Fock AIMD that can be conducted at DFT cost for medium-sized systems, (b) hybrid DFT condensed-phase studies at the cost of pure density functionals, (c) reduced cost on-the-fly large basis gas-phase AIMD and condensed-phase studies, (d) post-Hartree–Fock-level potential surfaces at DFT cost to obtain quantum nuclear effects, and (e) novel transfer machine learning protocols derived from these measures. Additionally, in previous work, the unifying strategy discussed here has been used to construct new quantum computing algorithms. Thus, we conclude that this reformulation is robust and accurate.
81 Capturing weak interactions in surface adsorbate systems at coupled cluster accuracy: a graph-theoretic molecular fragmentation approach improved through machine learning
Timothy C. Ricard, Xiao Zhu, and S. S. Iyengar, “Capturing weak interactions in surface adsorbate systems at coupled cluster accuracy: a graph-theoretic molecular fragmentation approach improved through machine learning”. J. Chem. Theory and Comput. 19, 8541 (2023).
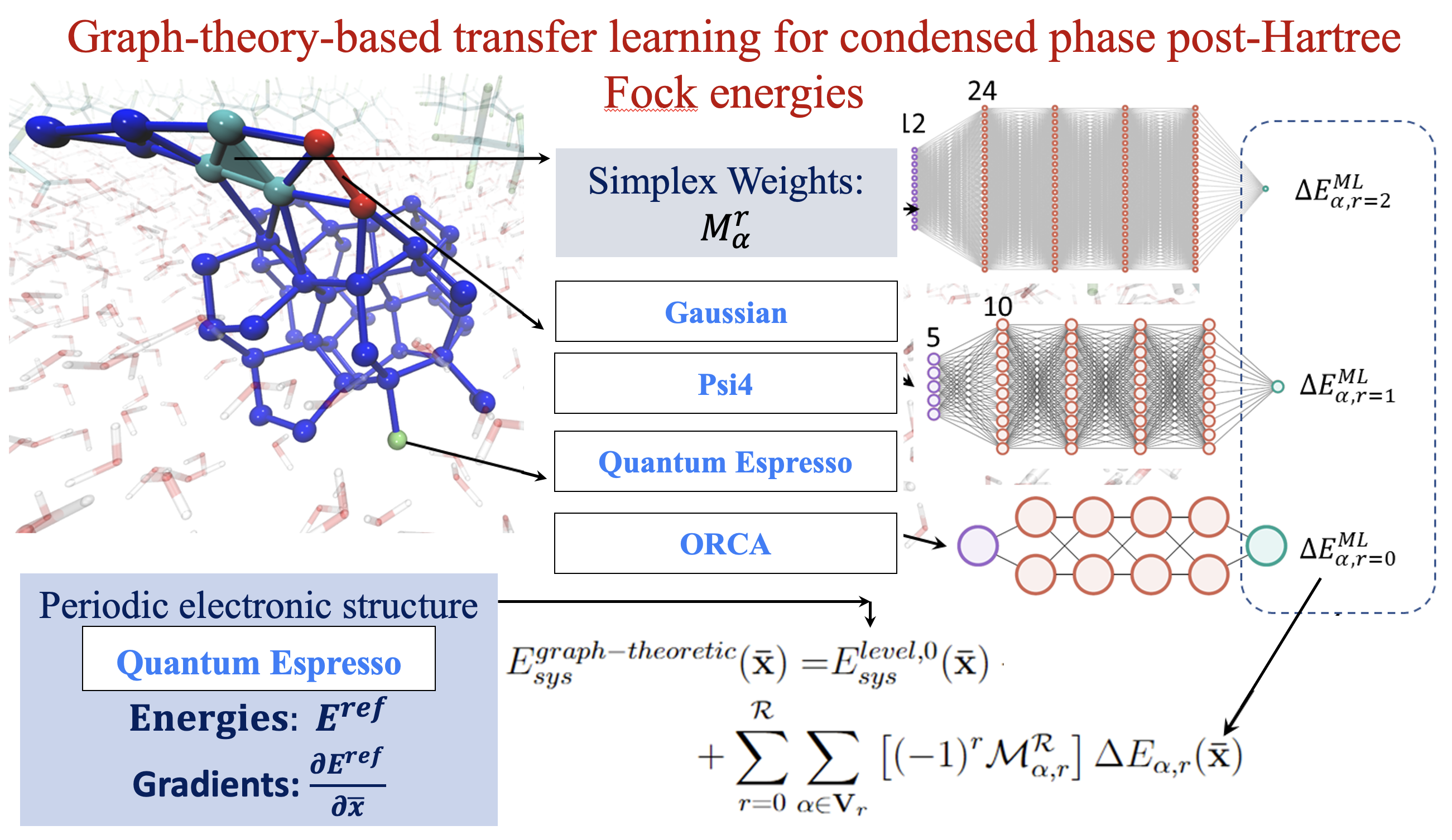 Summary: The accurate and efficient study of the interactions of organic matter with the surface of water is critical to a wide range of applications. For example, environmental studies have found that acidic polyfluorinated alkyl substances, especially perfluorooctanoic acid (PFOA), have spread throughout the environment and bioaccumulate into human populations residing near contaminated watersheds, leading to many systemic maladies. Thus, the study of the interactions of PFOA with water surfaces became important for the mitigation of their activity as pollutants and threats to public health. However, theoretical study of the interactions of such organic adsorbates on the surface of water, and their bulk concerted properties, often necessitates the use of ab initio methods to properly incorporate the long-range electronic properties that govern these extended systems. Notable theoretical treatments of “on-water” reactions thus far have employed hybrid DFT and semilocal DFT, but the interactions involved are weak interactions that may be best described using post-Hartree–Fock theory. Here, we aim to demonstrate the utility of a graph-theoretic approach to molecular fragmentation that accurately captures the critical “weak” interactions while maintaining an efficient ab initio treatment of the long-range periodic interactions that underpin the physics of extended systems. We apply this graph-theoretical treatment to study PFOA on the surface of water as a model system for the study of weak interactions seen in the wide range of surface interactions and reactions. The approach divides a system into a set of vertices, that are then connected through edges, faces, and higher order graph theoretic objects known as simplexes, to represent a collection of locally interacting subsystems. These subsystems are then used to construct ab initio molecular dynamics simulations and for computing multidimensional potential energy surfaces. To further improve the computational efficiency of our graph theoretic fragmentation method, we use a recently developed transfer learning protocol to construct the full system potential energy from a family of neural networks each designed to accurately model the behavior of individual simplexes. We use a unique multidimensional clustering algorithm, based on the k-means clustering methodology, to define our training space for each separate simplex. These models are used to extrapolate the energies for molecular dynamics trajectories at PFOA water interfaces, at less than one-tenth the cost as compared to a regular molecular fragmentation-based dynamics calculation with excellent agreement with couple cluster level of full system potential energies.
Summary: The accurate and efficient study of the interactions of organic matter with the surface of water is critical to a wide range of applications. For example, environmental studies have found that acidic polyfluorinated alkyl substances, especially perfluorooctanoic acid (PFOA), have spread throughout the environment and bioaccumulate into human populations residing near contaminated watersheds, leading to many systemic maladies. Thus, the study of the interactions of PFOA with water surfaces became important for the mitigation of their activity as pollutants and threats to public health. However, theoretical study of the interactions of such organic adsorbates on the surface of water, and their bulk concerted properties, often necessitates the use of ab initio methods to properly incorporate the long-range electronic properties that govern these extended systems. Notable theoretical treatments of “on-water” reactions thus far have employed hybrid DFT and semilocal DFT, but the interactions involved are weak interactions that may be best described using post-Hartree–Fock theory. Here, we aim to demonstrate the utility of a graph-theoretic approach to molecular fragmentation that accurately captures the critical “weak” interactions while maintaining an efficient ab initio treatment of the long-range periodic interactions that underpin the physics of extended systems. We apply this graph-theoretical treatment to study PFOA on the surface of water as a model system for the study of weak interactions seen in the wide range of surface interactions and reactions. The approach divides a system into a set of vertices, that are then connected through edges, faces, and higher order graph theoretic objects known as simplexes, to represent a collection of locally interacting subsystems. These subsystems are then used to construct ab initio molecular dynamics simulations and for computing multidimensional potential energy surfaces. To further improve the computational efficiency of our graph theoretic fragmentation method, we use a recently developed transfer learning protocol to construct the full system potential energy from a family of neural networks each designed to accurately model the behavior of individual simplexes. We use a unique multidimensional clustering algorithm, based on the k-means clustering methodology, to define our training space for each separate simplex. These models are used to extrapolate the energies for molecular dynamics trajectories at PFOA water interfaces, at less than one-tenth the cost as compared to a regular molecular fragmentation-based dynamics calculation with excellent agreement with couple cluster level of full system potential energies.
80 Graph-|Q⟩⟨C|: A Quantum algorithm with Reduced quantum circuit depth for electronic structure
S. S. Iyengar, Juncheng (Harry) Zhang, Timothy C. Ricard and Debadrita Saha, “Graph-|Q⟩⟨C|: A Quantum algorithm with Reduced quantum circuit depth for electronic structure”, J. Phys. Chem. A, 127, 9334 (2023).
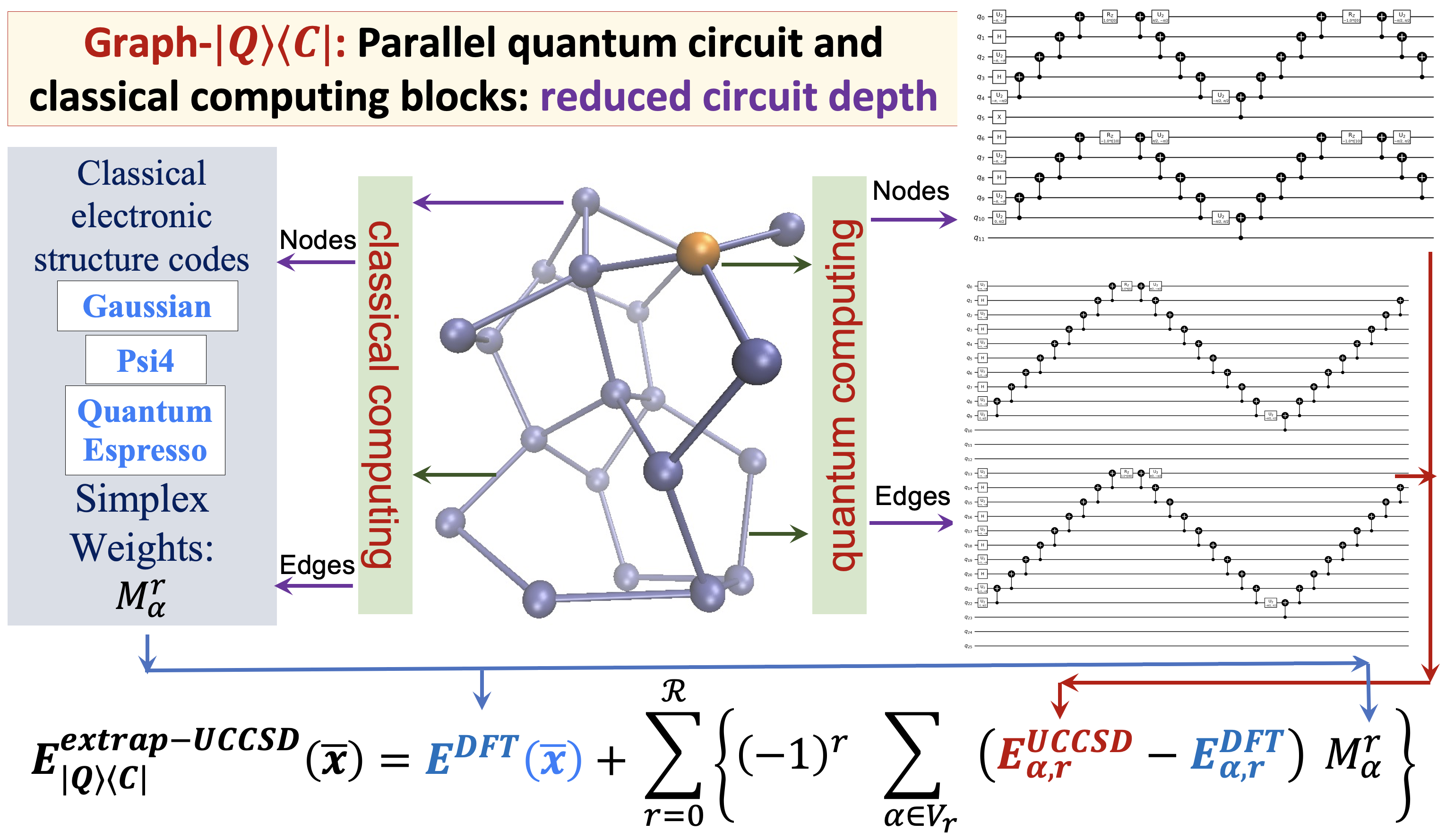 Summary: The accurate determination of chemical properties is known to have a critical impact on multiple fundamental chemical problems but is deeply hindered by the steep algebraic scaling of electron correlation calculations and the exponential scaling of quantum nuclear dynamics. With the advent of new quantum computing hardware and associated developments in creating new paradigms for quantum software, this avenue has been recognized as perhaps one way to address exponentially complex challenges in quantum chemistry and molecular dynamics. In this paper, we discuss a new approach to drastically reduce the quantum circuit depth (by several orders of magnitude) and help improve the accuracy in the quantum computation of electron correlation energies for large molecular systems. The method is derived from a graph-theoretic approach to molecular fragmentation and enables us to create a family of projection operators that decompose quantum circuits into separate unitary processes. Some of these processes can be treated on quantum hardware and others on classical hardware in a completely asynchronous and parallel fashion. Numerical benchmarks are provided through the computation of unitary coupled-cluster singles and doubles (UCCSD) energies for medium-sized protonated and neutral water clusters using the new quantum algorithms presented here.
Summary: The accurate determination of chemical properties is known to have a critical impact on multiple fundamental chemical problems but is deeply hindered by the steep algebraic scaling of electron correlation calculations and the exponential scaling of quantum nuclear dynamics. With the advent of new quantum computing hardware and associated developments in creating new paradigms for quantum software, this avenue has been recognized as perhaps one way to address exponentially complex challenges in quantum chemistry and molecular dynamics. In this paper, we discuss a new approach to drastically reduce the quantum circuit depth (by several orders of magnitude) and help improve the accuracy in the quantum computation of electron correlation energies for large molecular systems. The method is derived from a graph-theoretic approach to molecular fragmentation and enables us to create a family of projection operators that decompose quantum circuits into separate unitary processes. Some of these processes can be treated on quantum hardware and others on classical hardware in a completely asynchronous and parallel fashion. Numerical benchmarks are provided through the computation of unitary coupled-cluster singles and doubles (UCCSD) energies for medium-sized protonated and neutral water clusters using the new quantum algorithms presented here.
76 Quantum algorithms for the study of electronic structure and molecular dynamics: Novel computational protocols
S. S. Iyengar, Debadrita Saha, Anurag Dwivedi, Miguel Angel Lopez-Ruiz, Anup Kumar, Juncheng (Harry) Zhang, Timothy C. Ricard, Philip Richerme Amr Sabry “Quantum algorithms for the study of electronic structure and molecular dynamics: Novel computational protocols” In Comprehensive Computational Chemistry. In Press. (2023)
Summary: The accurate computational determination of chemical, materials, biological, and atmospheric properties has critical impact on a wide range of health and environmental problems, but is deeply limited by the computational scaling of quantum-mechanical methods. The complexity of quantum-chemical studies arises from the steep algebraic scaling of electron correlation methods, and the exponential scaling in studying nuclear dynamics and molecular flexibility. In this article we provide an overview of the challenges involved in performing accurate post-Hartree-Fock electronic structure and quantum nuclear dynamics calculations on quantum hardware. For electronic structure, we present a procedure to drastically reduce the depth of quantum circuits and improve the accuracy of results in computing post-Hartree-Fock electronic structure energies for large molecular systems. The method is based on molecular fragmentation where a molecular system is divided into overlapping fragments through a graph theoretic procedure. This allows us to create a set of projection operators that decompose the unitary evolution of the full system into separate sets of unitary processes, some of which can be treated on quantum hardware and others on classical hardware. Thus, we develop a procedure for electronic structure that can be asynchronously spawned onto a potentially large ensemble of classical and quantum hardware systems. We also discuss a framework which allows for the solution of quantum chemical nuclear dynamics by mapping these to quantum spin-lattice simulators. This mapping procedure allows us to determine the local fields and spin-spin couplings needed to identically match the molecular and spin-lattice Hamiltonians and hence the resultant dynamics.
74 Graph-theoretic Molecular fragmentation for potential surfaces leads naturally to a tensor network form and allows accurate and efficient quantum nuclear dynamics
A. Kumar, N. DeGregorio T. C. Ricard, and S. S. Iyengar, “Graph-theoretic Molecular fragmentation for potential surfaces leads naturally to a tensor network form and allows accurate and efficient quantum nuclear dynamics”. J. Chem. Theory and Comput. 18, 7243 (2022).
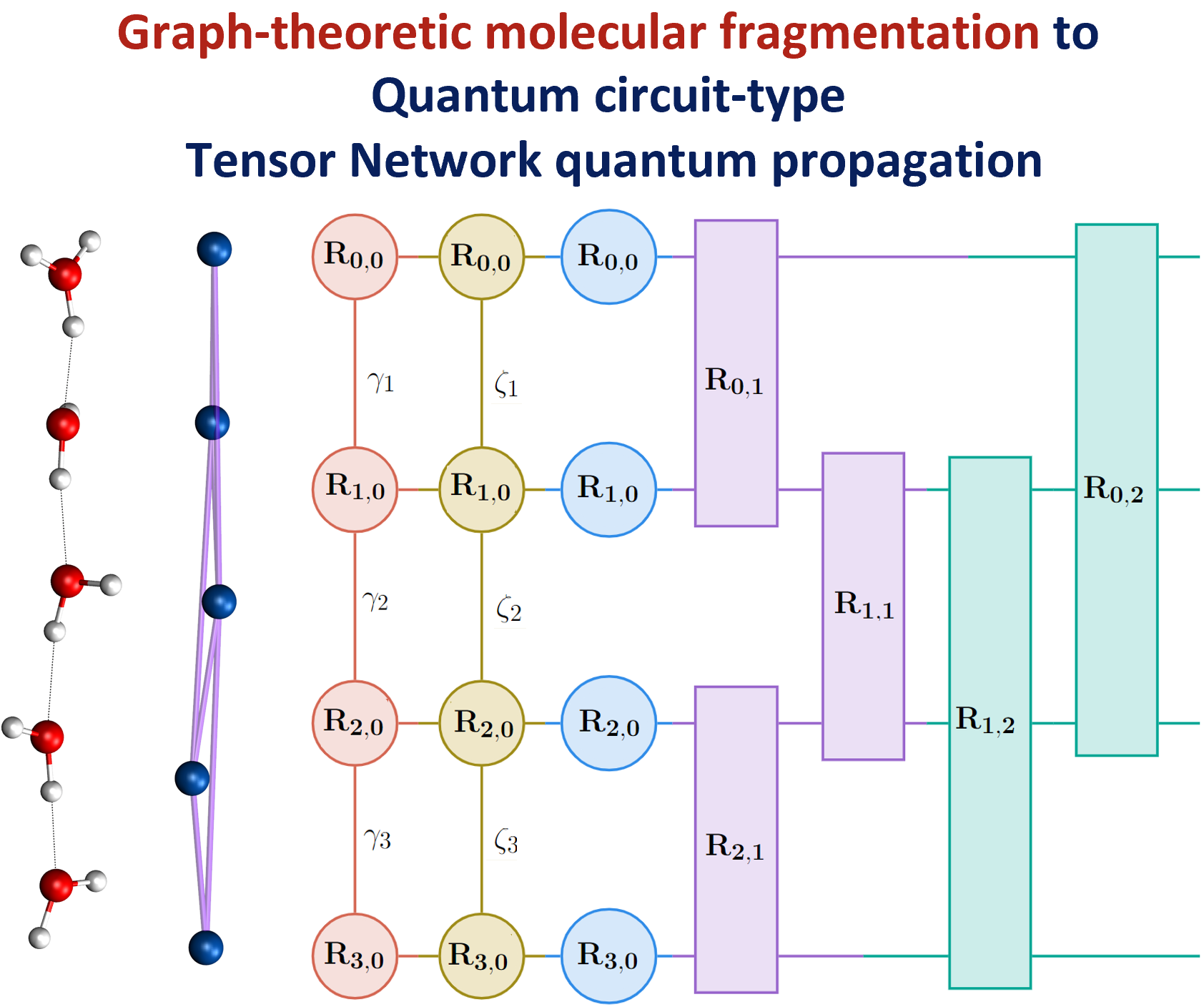 Summary: Molecular fragmentation methods have revolutionized quantum chemistry. Here, we use a graph-theoretically generated molecular fragmentation method, to obtain accurate and efficient representations for multidimensional potential energy surfaces and the quantum time-evolution operator, which plays a critical role in quantum chemical dynamics. In doing so, we find that the graph-theoretic fragmentation approach naturally reduces the potential portion of the time-evolution operator into a tensor network that contains a stream of coupled lower-dimensional propagation steps to potentially achieve quantum dynamics with reduced complexity. Furthermore, the fragmentation approach used here has previously been shown to allow accurate and efficient computation of post- Hartree−Fock electronic potential energy surfaces, which in many cases has been shown to be at density functional theory cost. Thus, by combining the advantages of molecular fragmentation with the tensor network formalism, the approach yields an on-the-fly quantum dynamics scheme where both the electronic potential calculation and nuclear propagation portion are enormously simplified through a single stroke. The method is demonstrated by computing approximations to the propagator and to potential surfaces for a set of coupled nuclear dimensions within a protonated water wire problem exhibiting the Grotthuss mechanism of proton transport. In all cases, our approach has been shown to reduce the complexity of representing the quantum propagator, and by extension action of the propagator on an initial wavepacket, by several orders, with minimal loss in accuracy.
Summary: Molecular fragmentation methods have revolutionized quantum chemistry. Here, we use a graph-theoretically generated molecular fragmentation method, to obtain accurate and efficient representations for multidimensional potential energy surfaces and the quantum time-evolution operator, which plays a critical role in quantum chemical dynamics. In doing so, we find that the graph-theoretic fragmentation approach naturally reduces the potential portion of the time-evolution operator into a tensor network that contains a stream of coupled lower-dimensional propagation steps to potentially achieve quantum dynamics with reduced complexity. Furthermore, the fragmentation approach used here has previously been shown to allow accurate and efficient computation of post- Hartree−Fock electronic potential energy surfaces, which in many cases has been shown to be at density functional theory cost. Thus, by combining the advantages of molecular fragmentation with the tensor network formalism, the approach yields an on-the-fly quantum dynamics scheme where both the electronic potential calculation and nuclear propagation portion are enormously simplified through a single stroke. The method is demonstrated by computing approximations to the propagator and to potential surfaces for a set of coupled nuclear dimensions within a protonated water wire problem exhibiting the Grotthuss mechanism of proton transport. In all cases, our approach has been shown to reduce the complexity of representing the quantum propagator, and by extension action of the propagator on an initial wavepacket, by several orders, with minimal loss in accuracy.
73 Graph theoretic molecular fragmentation methods for electronic potential energy surfaces augmented by machine learning
Xiao Zhu and S. S. Iyengar, “Graph Theoretic Molecular Fragmentation for Multidimensional Potential Energy Surfaces Yield an Adaptive and General Transfer Machine Learning Protocol”, Chem. Theory and Comput. 18, 5125 (2022).
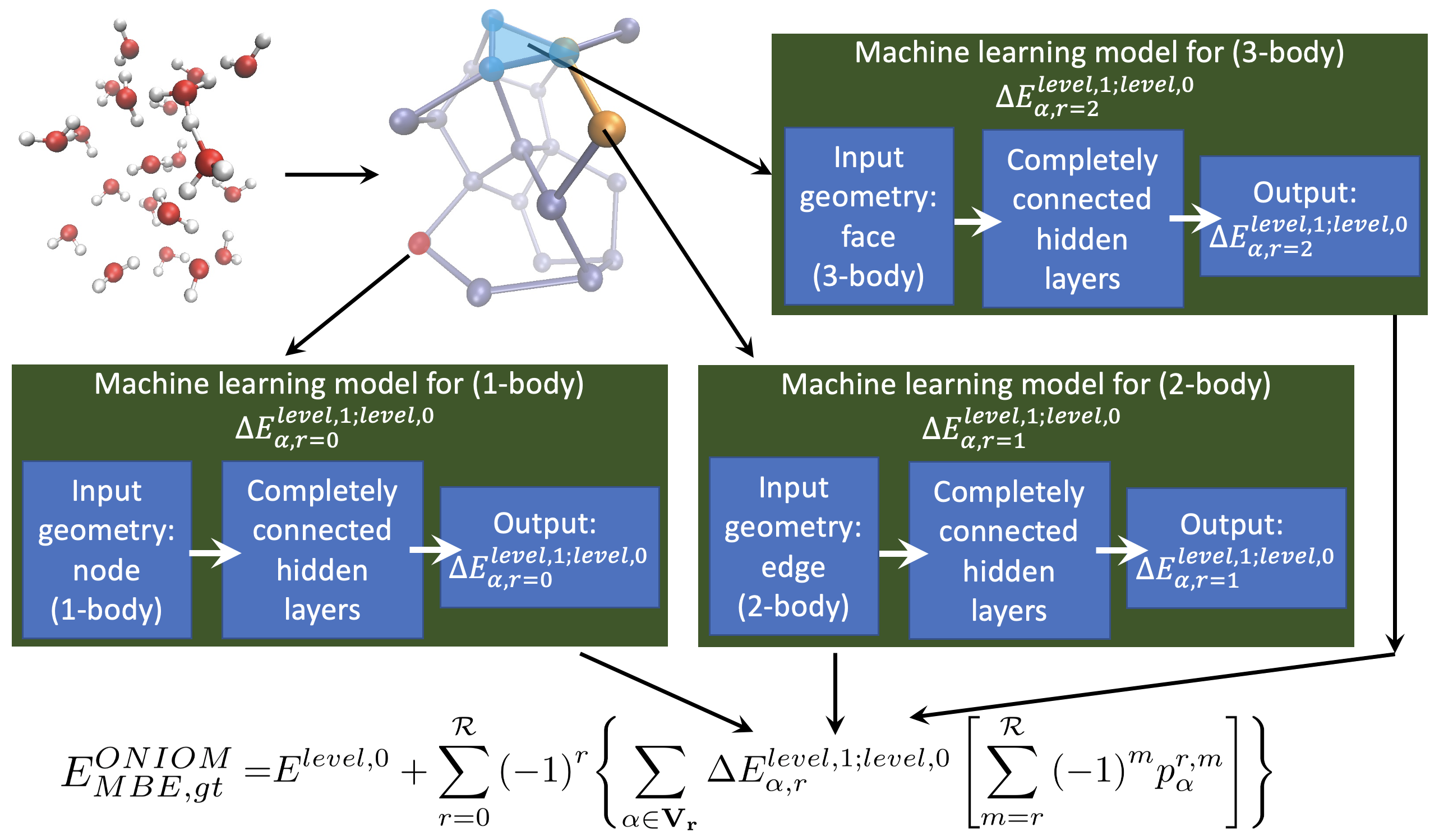 Summary: Over a series of publications we have introduced a graph-theoretic description for molecular fragmentation. Here, a system is divided into a set of nodes, or vertices, that are then connected through edges, faces, and higher-order simplexes to represent a collection of spatially overlapping and locally interacting subsystems. Each such subsystem is treated at two levels of electronic structure theory, and the result is used to construct many-body expansions that are then embedded within an ONIOM-scheme. These expansions converge rapidly with many-body order (or graphical rank) of subsystems and have been previously used for ab initio molecular dynamics (AIMD) calculations and for computing multidimensional potential energy surfaces. Specifically, in all these cases we have shown that CCSD and MP2 level AIMD trajectories and potential surfaces may be obtained at density functional theory cost. The approach has been demonstrated for gas-phase studies, for condensed phase electronic structure, and also for basis set extrapolation-based AIMD. Recently, this approach has also been used to derive new quantum-computing algorithms that enormously reduce the quantum circuit depth in a circuit-based computation of correlated electronic structure. In this publication, we introduce (a) a family of neural networks that act in parallel to represent, efficiently, the post-Hartree−Fock electronic structure energy contributions for all simplexes (fragments), and (b) a new k-means-based tessellation strategy to glean training data for high-dimensional molecular spaces and minimize the extent of training needed to construct this family of neural networks. The approach is particularly useful when coupled cluster accuracy is desired and when fragment sizes grow in order to capture nonlocal interactions accurately. The unique multidimensional k-means tessellation/clustering algorithm used to determine our training data for all fragments is shown to be extremely efficient and reduces the needed training to only 10% of data for all fragments to obtain accurate neural networks for each fragment. These fully connected dense neural networks are then used to extrapolate the potential energy surface for all molecular fragments, and these are then combined as per our graph-theoretic procedure to transfer the learning process to a full system energy for the entire AIMD trajectory at less than one-tenth the cost as compared to a regular fragmentation-based AIMD calculation.
Summary: Over a series of publications we have introduced a graph-theoretic description for molecular fragmentation. Here, a system is divided into a set of nodes, or vertices, that are then connected through edges, faces, and higher-order simplexes to represent a collection of spatially overlapping and locally interacting subsystems. Each such subsystem is treated at two levels of electronic structure theory, and the result is used to construct many-body expansions that are then embedded within an ONIOM-scheme. These expansions converge rapidly with many-body order (or graphical rank) of subsystems and have been previously used for ab initio molecular dynamics (AIMD) calculations and for computing multidimensional potential energy surfaces. Specifically, in all these cases we have shown that CCSD and MP2 level AIMD trajectories and potential surfaces may be obtained at density functional theory cost. The approach has been demonstrated for gas-phase studies, for condensed phase electronic structure, and also for basis set extrapolation-based AIMD. Recently, this approach has also been used to derive new quantum-computing algorithms that enormously reduce the quantum circuit depth in a circuit-based computation of correlated electronic structure. In this publication, we introduce (a) a family of neural networks that act in parallel to represent, efficiently, the post-Hartree−Fock electronic structure energy contributions for all simplexes (fragments), and (b) a new k-means-based tessellation strategy to glean training data for high-dimensional molecular spaces and minimize the extent of training needed to construct this family of neural networks. The approach is particularly useful when coupled cluster accuracy is desired and when fragment sizes grow in order to capture nonlocal interactions accurately. The unique multidimensional k-means tessellation/clustering algorithm used to determine our training data for all fragments is shown to be extremely efficient and reduces the needed training to only 10% of data for all fragments to obtain accurate neural networks for each fragment. These fully connected dense neural networks are then used to extrapolate the potential energy surface for all molecular fragments, and these are then combined as per our graph-theoretic procedure to transfer the learning process to a full system energy for the entire AIMD trajectory at less than one-tenth the cost as compared to a regular fragmentation-based AIMD calculation.
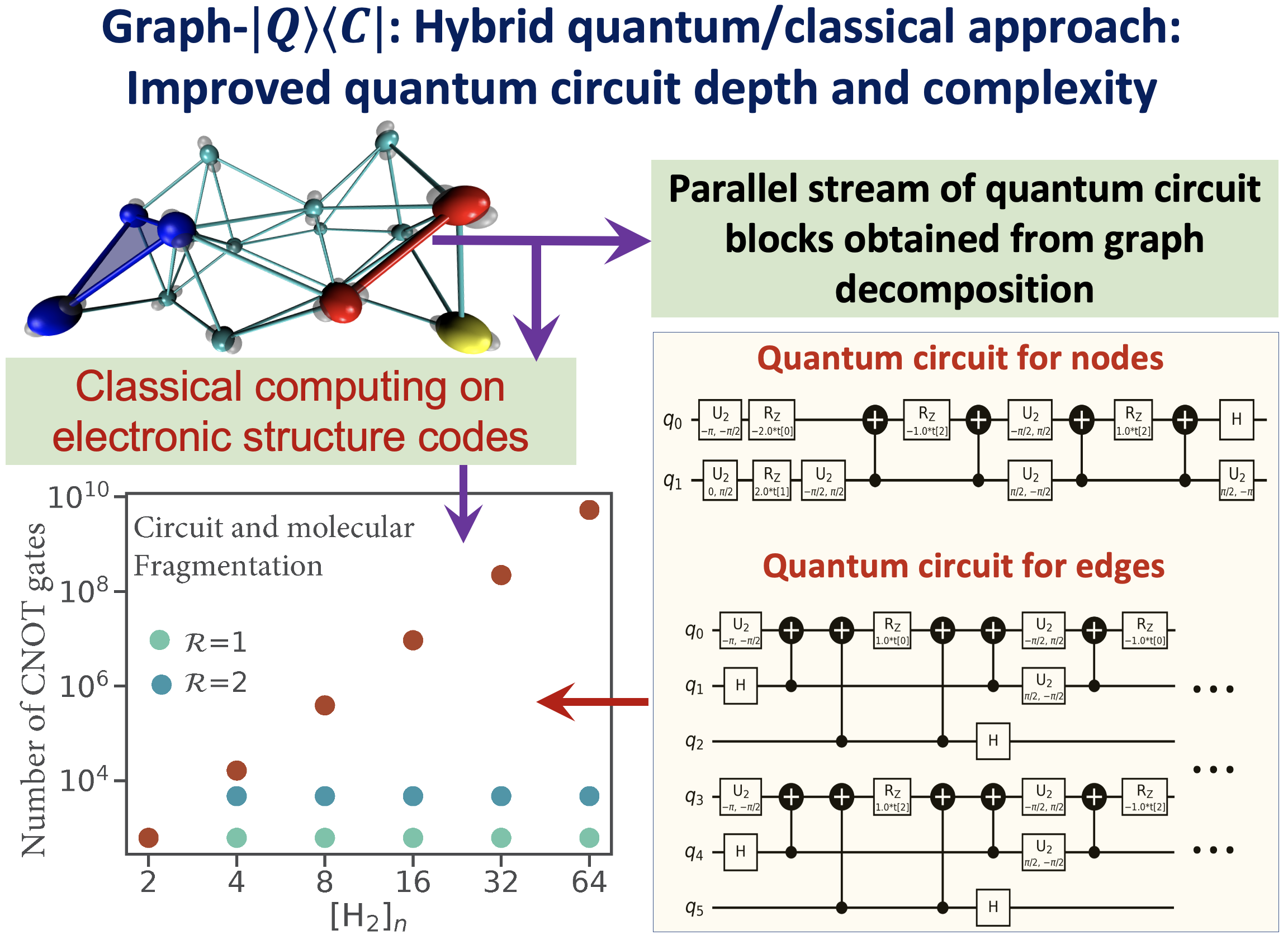
72 Graph-|Q⟩⟨C|, a Graph-Based Quantum/Classical Algorithm for Efficient Electronic Structure on Hybrid Quantum/Classical Hardware Systems: Improved Quantum Circuit Depth Performance
 Juncheng Harry Zhang, and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 18, 2885 (2022).
Juncheng Harry Zhang, and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 18, 2885 (2022).
Summary: Recently, multiple quantum computing technologies have emerged as potential alternative computational platforms to address complex computational challenges. Additionally, algorithms to approximate electron correlation problems, for small molecular systems, and quantum nuclear dynamics problems have been implemented on quantum hardware devices. However, application of standard quantum circuit models to treat electronic structure problems leads to a rapid increase in the circuit depth and the number of quantum gates. This contributes greatly to the accumulated error during quantum propagation. This paper outlines a new hybrid quantum+classical algorithm based on a graph-theoretic approach to molecular fragmentation and is geared toward performing electron correlation calculations, potentially on an ensemble of quantum and classical hardware systems. The algorithm studied here is referred to as the “Graph-|Q⟩⟨C|” algorithm since it contains an independent set of classical and quantum algorithmic components inside a single umbrella. That is, the overall computational workload is partitioned, through graph theory based on computational complexity analysis, into (a) classical computing sections that are carried out on traditional classical electronic structure packages, and (b) quantum computing sections that are carried out using quantum circuit models. Furthermore, the Graph-|Q⟩⟨C| algorithm is quantum hardware-agnostic and is developed with the goal to be implemented on all quantum hardware technologies, and, in fact, is designed to be used on an ensemble of such quantum hardware systems for any given calculation. In essence, our Graph-|Q⟩⟨C| algorithm yields a new approach that reduces the required quantum circuit depth, the number of quantum gates, and the number of CNOT gates (by several orders of magnitude) that contribute to error accumulation, through a graph-theory-based projection operator formalism. Thus, given this reduction, our algorithm, potentially improves the quantum algorithmic efficiency, provides a new avenue for quantum resource management, and also reduces the accumulation of errors during the demonstrated electronic structure calculations on quantum hardware. Given the limitations of quantum circuit gate fidelities within the gate model, this algorithm, we expect, will become a central piece in the quantum/classical computing of chemical systems.
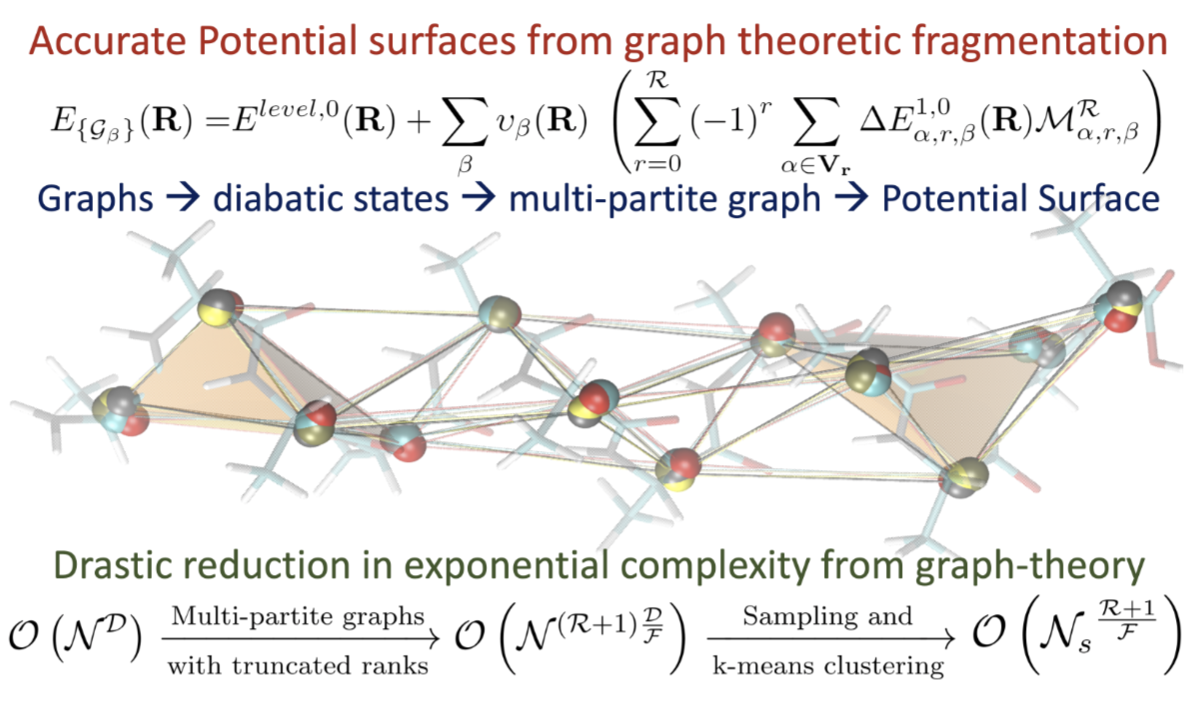
70 Graph-theory based molecular fragmentation for efficient and accurate potential surface calculations in multiple dimensions
 Anup Kumar, Nicole DeGregorio and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 17, 6671 (2021).
Anup Kumar, Nicole DeGregorio and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 17, 6671 (2021).
Summary: The accurate and efficient study of electronic structure and nuclear dynamics is at the core of multiple problems that are critical to materials, biological, and atmospheric research. However, these studies are deeply affected by the steep, polynomial scaling cost of electronic structure, and potentially exponential scaling of quantum nuclear dynamics. In this publication, we present a multi-topology molecular fragmentation approach, based on graph theory, to calculate multi-dimensional potential energy surfaces in agreement with post-Hartree-Fock levels of theory, but at DFT cost.
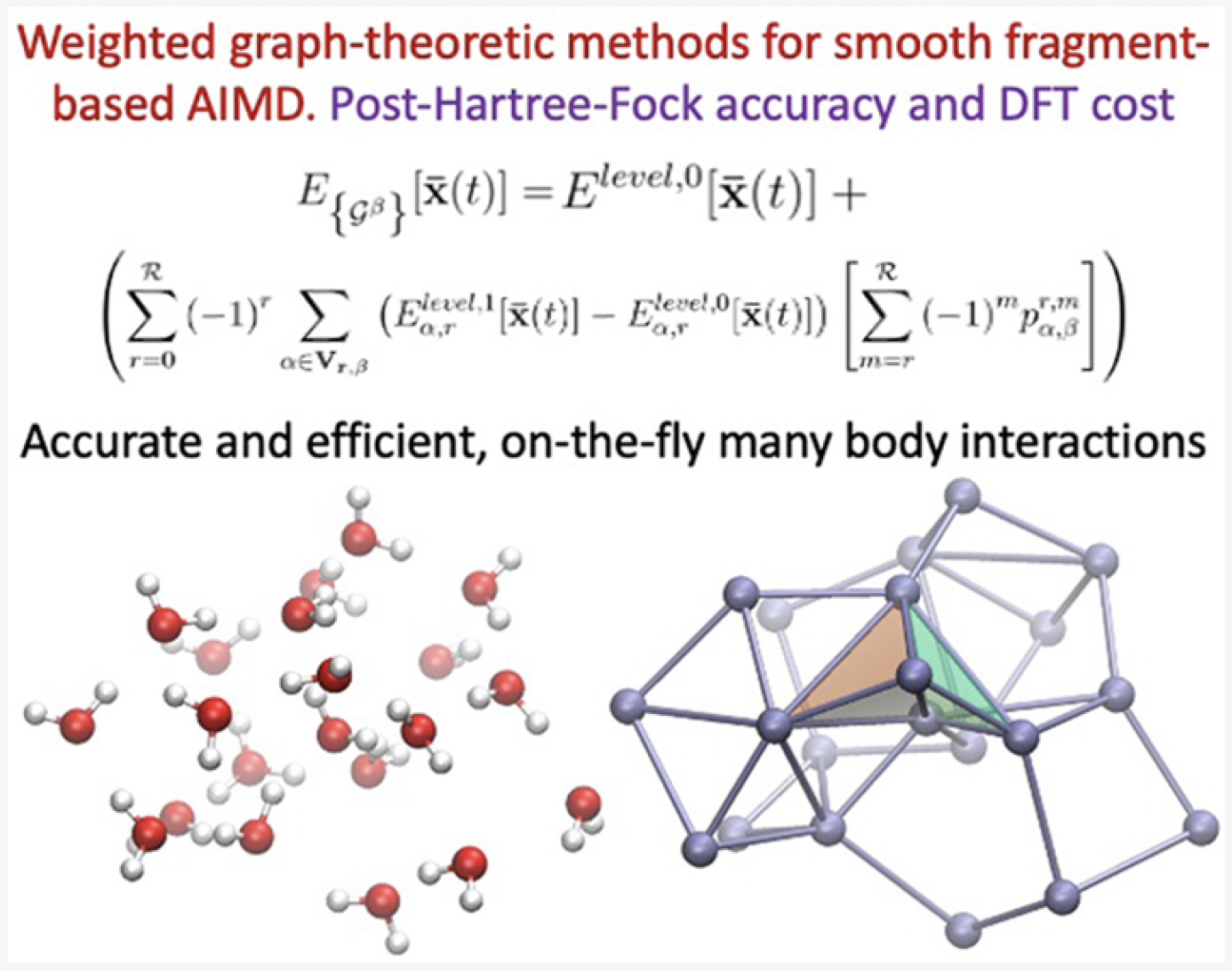
69 Weighted-Graph-Theoretic Methods for Many-Body Corrections within ONIOM: Smooth AIMD and the Role of High-Order ManyBody Terms
 Juncheng Harry Zhang, Timothy C. Ricard, Cody Haycraft, and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 17, 2672 (2021).
Juncheng Harry Zhang, Timothy C. Ricard, Cody Haycraft, and Srinivasan S. Iyengar, J. Chem. Theory and Comput. 17, 2672 (2021).
Summary: We present a weighted-graph-theoretic approach to adaptively compute contributions from many-body approximations for smooth and accurate post-Hartree−Fock (pHF) ab initio molecular dynamics (AIMD) of highly fluxional chemical systems
68 An efficient and accurate approach to estimate hybrid functional and large basis set contributions to condensed phase systems and molecule-surface interactions
![]() T. C. Ricard and S. S. Iyengar, J. Chem. Theory and Comput. 16, 4790 (2020). (Supporting Information)
Summary: We present an efficient approach to perform accurate hybrid DFT electronic structure calculations for condensed phase systems, at pure DFT cost.
T. C. Ricard and S. S. Iyengar, J. Chem. Theory and Comput. 16, 4790 (2020). (Supporting Information)
Summary: We present an efficient approach to perform accurate hybrid DFT electronic structure calculations for condensed phase systems, at pure DFT cost.
67 Embedded, graph-theoretically defined many-body approximations for wavefunction-in-DFT and DFT-in-DFT: applications to gas- and condensed-phase AIMD, and potential surfaces for quantum nuclear effects
Timothy C. Ricard, Anup Kumar and Srinivasan S. Iyengar, Int. J. Quant. Chem. In Press. . Special issue on “Quantum Embedding Electronic Structure Methods”. 120, e26244 (2020). Summary: We discuss a graph theoretic approach to adaptively compute many-body-approximations in an efficient manner to perform (a) accurate post-Hartree-Fock AIMD at DFT cost for medium to large sized molecular clusters, (b) hybrid DFT electronic structure calculations for condensed phase simulations at the cost of pure density functionals, (c) reduced cost on-the-fly basis extrapolation for gas-phase AIMD and condensed phase studies, and (d) similar basis-set extrapolations for condensed phase calculations, and (e) accurate post-Hartree-Fock level potential energy surfaces at DFT cost for quantum nuclear effects.
66 Fragment-based electronic structure for potential energy surfaces using a superposition of fragmentation topologies
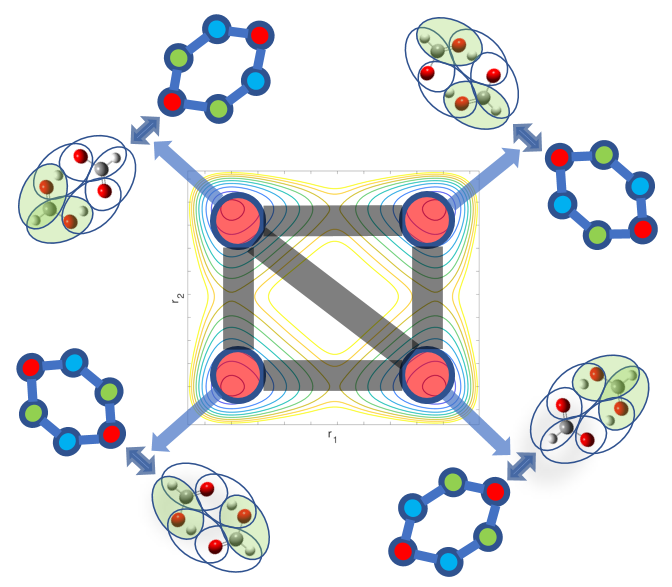 A. Kumar and S. S. Iyengar, "Fragment-based electronic structure for potential energy surfaces using a superposition of fragmentation topologies", J. Chem. Theory and Comput. 15, 5769 (2019).
A. Kumar and S. S. Iyengar, "Fragment-based electronic structure for potential energy surfaces using a superposition of fragmentation topologies", J. Chem. Theory and Comput. 15, 5769 (2019).
Summary: Molecular fragmentation methods developed by us and by several groups have made it possible to perform accurate electronic structure calculations in large systems. However, problems exist with these ideas when are applied to molecular potential surface calculations where fragments may change as the nuclei move. We develop a Fragment-Based electronic structure method which adaptively utilizes multiple graphical representations for a given molecular system and use these to compute smooth potential energy surfaces in an efficent manner. Graph theory based representations are used to efficiently compute and represent the local interactions between coarse grain units of molecules leading to post-Hartree-Fock accurate results at DFT cost. Benchmarks are provided on protonated waterwire systems ubiquitous in numerous biological ion channels and in material applications.
62 Efficiently capturing weak interactions in ab initio molecular dynamics with on the fly basis set extrapolation
T. C. Ricard and S. S. Iyengar, "Efficiently capturing weak interactions in ab initio molecular dynamics with on the fly basis set extrapolation", J. Chem. Theory and Comput. 14, 5535 (2018).
60 Adaptive, geometric networks for efficient coarse-grained ab initio molecular dynamics with post-Hartree-Fock accuracy
T. C. Ricard, C. Haycraft, and S. S. Iyengar, "Adaptive, geometric networks for efficient coarse-grained ab initio molecular dynamics with post-Hartree-Fock accuracy", J. Chem. Theory and Comput. 14, 30 (2018).
56 Efficient, 'On-the-fly', Born-Oppenheimer and Car-Parrinello-type dynamics with coupled cluster (CCSD) accuracy through fragment based electronic structure
C. Haycraft, J. Li and S. S. Iyengar, "Efficient, 'On-the-fly', Born-Oppenheimer and Car-Parrinello-type dynamics with coupled cluster (CCSD) accuracy through fragment based electronic structure" J. Chem. Theory and Comput. 13, 1885 (2017). Supporting Information: Click here
54 Hybrid extended Lagrangian, post-Hartree-Fock Born-Oppenheimer ab initio molecular dynamics with fragment-based electronic structure
J. Li, C. Haycraft and S. S. Iyengar "Hybrid extended Lagrangian, post-Hartree-Fock Born-Oppenheimer ab initio molecular dynamics with fragment-based electronic structure". J. Chem. Theory and Comput. 12, 2493 (2016).
53 Ab initio molecular dynamics using recursive, spatially separated, overlapping model subsystems mixed within an ONIOM based fragmentation energy extrapolation technique
J. Li and S. S. Iyengar "Ab initio molecular dynamics using recursive, spatially separated, overlapping model subsystems mixed within an ONIOM based fragmentation energy extrapolation technique". J. Chem. Theory and Comput. 11, 3978 (2015).
2 Inclusion of molecular flexibility in Partition Coefficient calculations on oligopeptides using stochastic sampling
N. G. J. Richards, P. B. Williams, S. S. Iyengar, Proceedings of the twenty-seventh Hawaii International Conference on System Sciences 203 (1994).
 The College of Arts
The College of Arts